

Unraveling the glyco-immunity nexus in pancreatic cancer
- PMID: 40759948
- PMCID: PMC12320354
- DOI: 10.1186/s12943-025-02417-4
Unraveling the glyco-immunity nexus in pancreatic cancer
Abstract
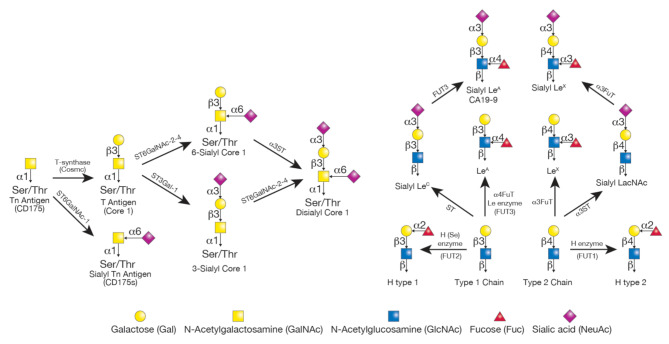
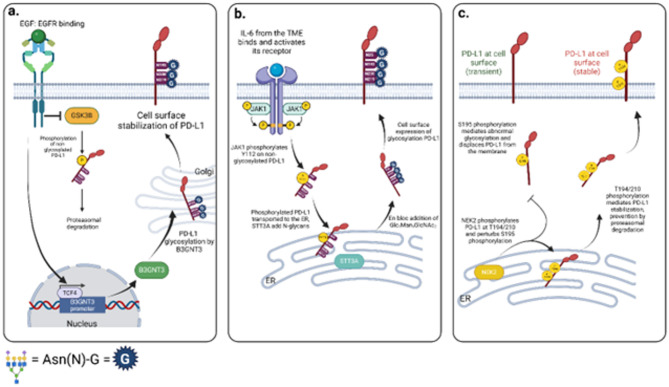
Pancreatic ductal adenocarcinoma (PDAC) is an extremely aggressive disease, and standard of care therapies have failed to yield significant clinical benefit, with invasive surgery being the only curative treatment for patients with early-stage disease. Tumor-associated glycans in pancreatic cancer have direct effects on the survival and propagation of the tumor proper and contribute to an immunosuppressed tumor microenvironment. The existence of a “tumor glycocode” in PDAC and the role of hypersialylation in this cancer have been hugely underscored. Through this initial understanding, significant strides have been made in the field of glycosylation-mediated immune regulation, uncovering glyco-immune checkpoints that facilitate tumor progression in PDAC and other malignancies. Here, we describe the specific roles of glycan-binding proteins, such as C-type lectin receptors, Siglecs, and Galectins, in generating and promoting immunosuppression, exacerbating survival outcomes, and dampening therapeutic efficacy. We illustrate the scale of glycan-mediated regulation of homeostatic immune responses and how cancer glycans facilitate dampened anti-tumor immunity through the major histocompatibility complex (MHC) and the enhanced expression of immune checkpoints, such as PD-L1 and CTLA-4. A wide array of glycan-targeted therapies against PDAC in the clinic, including monoclonal antibodies, enzymes, and vaccines, has been described. With the help of new glycosylation signatures identified and techniques that allow us to reach single-cell resolution, we can target glycans and generate strategies to activate the immune system against PDAC.
Keywords: Immune signaling; Pancreatic cancer; Therapies; Tumor glycosylation; Tumor microenvironment.
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures



References
-
- Levene PA, Alsberg CL. The cleavage products of vitellin. J Biol Chem. 1906;2:127–33. 10.1016/S0021-9258(17)46054-6 - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
