

Insights into targeted ferroptosis in mechanisms, biology, and role of Alzheimer's disease: an update
- PMID: 40761698
- PMCID: PMC12319056
- DOI: 10.3389/fnagi.2025.1587986
Insights into targeted ferroptosis in mechanisms, biology, and role of Alzheimer's disease: an update
Abstract
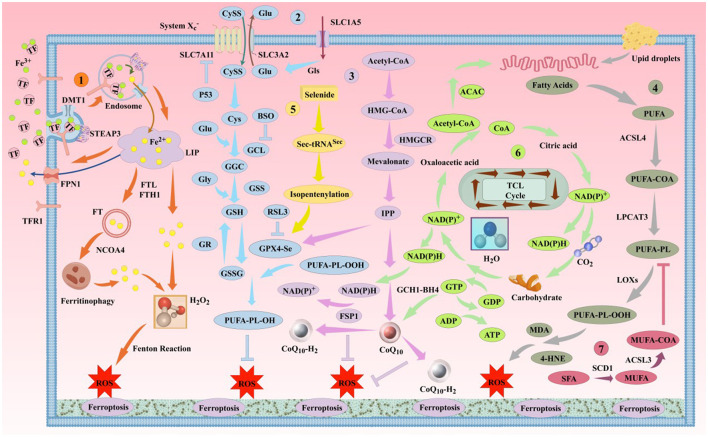
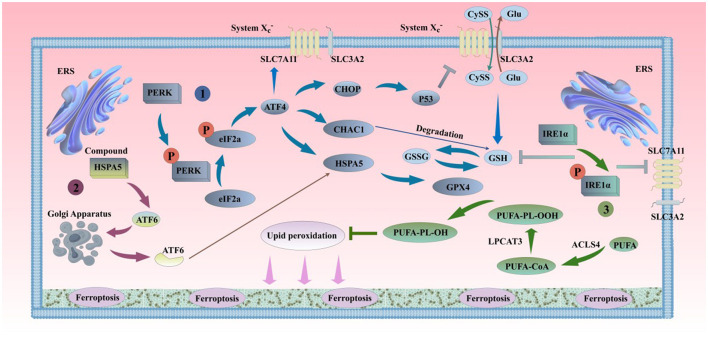
Ferroptosis is a newly discovered form of programmed cell death, primarily caused by an imbalance between iron-dependent oxidative damage and antioxidant defense mechanisms within the cell. It differs from previously reported forms of cell death, such as apoptosis, necrosis, and autophagy, in terms of morphology, biochemistry, and genetics. Alzheimer's disease (AD) is the most common neurodegenerative disorder, characterized by pathological features including neurofibrillary tangles (NFTs), senile plaques (SPs), and abnormal iron deposition, suggesting that ferroptosis may be involved in its disease progression. Although recent studies have made significant progress, the mechanisms underlying neuronal ferroptosis in AD remain incompletely understood. This review, based on elucidating the process and regulatory mechanisms of cellular ferroptosis, explores, and supplements the correlation between iron overload and redox imbalance with the main pathological mechanisms of AD, providing new insights for the treatment of AD and the development of new drugs.
Keywords: Alzheimer's disease; biology; ferroptosis; mechanisms; update.
Copyright © 2025 Zhou, Li, Wu, Wang, Cheng, Yang, Gao and Zhu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures







References
Publication types
LinkOut - more resources
Full Text Sources