

Design of cross-reactive antigens with machine learning and high-throughput experimental evaluation
- PMID: 40761757
- PMCID: PMC12319226
- DOI: 10.3389/fbinf.2025.1580967
Design of cross-reactive antigens with machine learning and high-throughput experimental evaluation
Abstract
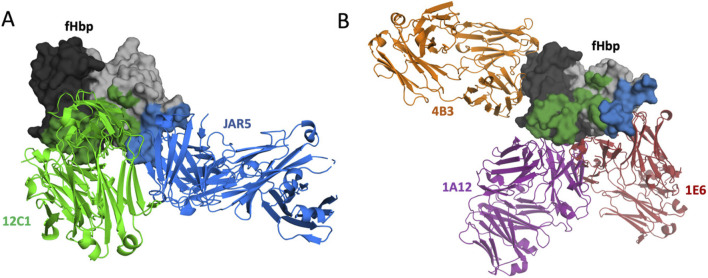
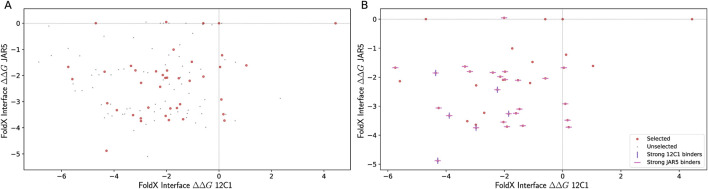
Selecting an optimal antigen is a crucial step in vaccine development, significantly influencing both the vaccine's effectiveness and the breadth of protection it provides. High antigen sequence variability, as seen in pathogens like rhinovirus, HIV, influenza virus, complicates the design of a single cross-protective antigen. Consequently, vaccination with a single antigen molecule often confers protection against only a single variant. In this study, machine learning methods were applied to the design of factor H binding protein (fHbp), an antigen from the bacterial pathogen Neisseria meningitidis. The vast number of potential antigen mutants presents a significant challenge for improving fHbp antigenicity. Moreover, limited data on antigen-antibody binding in public databases constrains the training of machine learning models. To address these challenges, we used computational models to predict fHbp properties and machine learning was applied to select both the most promising and informative mutants using a Gaussian process (GP) model. These mutants were experimentally evaluated to both confirm promising leads and refine the machine learning model for future iterations. In our current model, mutants were designed that enabled the transfer of fHbp v1.1 specific conformational epitopes onto fHbp v3.28, while maintaining binding to overlapping cross-reactive epitopes. The top mutant identified underwent biophysical and x-ray crystallographic characterization to confirm that the overall structure of fHbp was maintained throughout this epitope engineering experiment. The integrated strategy presented here could form the basis of a next-generation, iterative antigen design platform, potentially accelerating the development of new broadly protective vaccines.
Keywords: AI; ML; Neisseria meningitidis; antibody; antigen; protein engineering; protein structure; vaccine.
Copyright © 2025 Chesterman, Desautels, Sierra, Arrildt, Zemla, Lau, Sundaram, Laliberte, Chen, Ruby, Mednikov, Bertholet, Yu, Luisi, Malito, Mallett, Bottomley, van den Berg and Faissol.
Conflict of interest statement
Authors CC, L-JS, KA, JL, LC, AR, MM, SB, DY, KL, EM CM, MB, RB were employed by GSK and may have received GSK shares as part of their renumeration package. Several authors are listed as inventors on patents owned by GSK. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Immunogenicity and seroefficacy of pneumococcal conjugate vaccines: a systematic review and network meta-analysis.Health Technol Assess. 2024 Jul;28(34):1-109. doi: 10.3310/YWHA3079. Health Technol Assess. 2024. PMID: 39046101 Free PMC article.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2021 Apr 19;4(4):CD011535. doi: 10.1002/14651858.CD011535.pub4. Cochrane Database Syst Rev. 2021. Update in: Cochrane Database Syst Rev. 2022 May 23;5:CD011535. doi: 10.1002/14651858.CD011535.pub5. PMID: 33871055 Free PMC article. Updated.
-
Does the Presence of Missing Data Affect the Performance of the SORG Machine-learning Algorithm for Patients With Spinal Metastasis? Development of an Internet Application Algorithm.Clin Orthop Relat Res. 2024 Jan 1;482(1):143-157. doi: 10.1097/CORR.0000000000002706. Epub 2023 Jun 12. Clin Orthop Relat Res. 2024. PMID: 37306629 Free PMC article.
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
-
Stabilizing machine learning for reproducible and explainable results: A novel validation approach to subject-specific insights.Comput Methods Programs Biomed. 2025 Sep;269:108899. doi: 10.1016/j.cmpb.2025.108899. Epub 2025 Jun 21. Comput Methods Programs Biomed. 2025. PMID: 40570739
References
-
- Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., et al. (2015). GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25. 10.1016/j.softx.2015.06.001 - DOI
-
- Bachas S., Rakocevic G., Spencer D., Sastry A. V., Haile R., Sutton J. M., et al. (2022). Antibody optimization enabled by artificial intelligence predictions of binding affinity and naturalness. bioRxiv. 10.1101/2022.08.16.504181 - DOI
LinkOut - more resources
Full Text Sources