

14-3-3 binding maintains the Parkinson's associated kinase LRRK2 in an inactive state
- PMID: 40764514
- PMCID: PMC12325948
- DOI: 10.1038/s41467-025-62337-1
14-3-3 binding maintains the Parkinson's associated kinase LRRK2 in an inactive state
Abstract
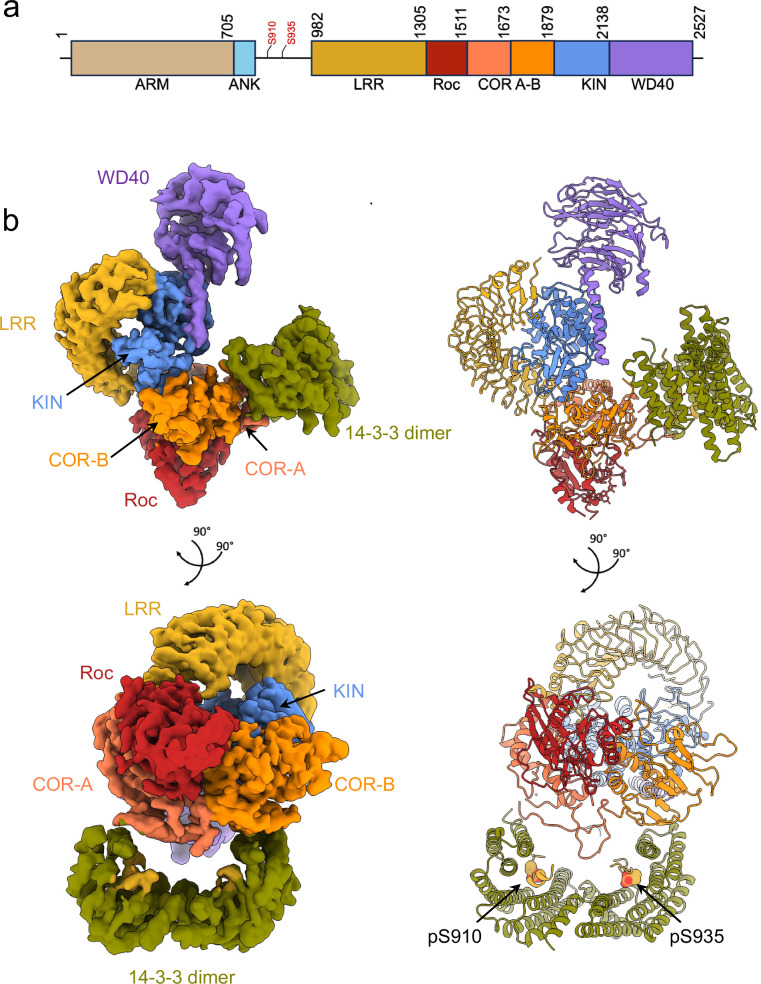
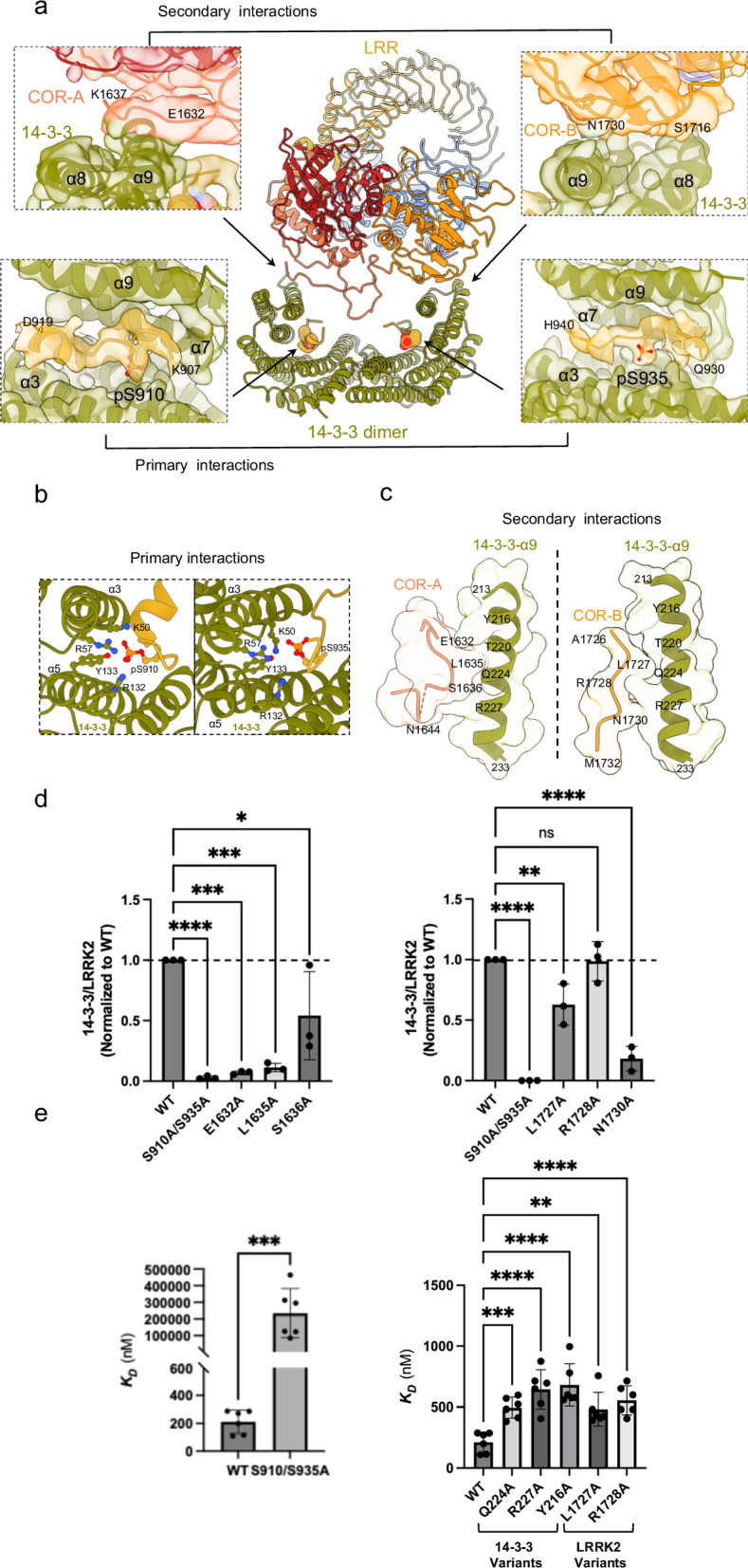
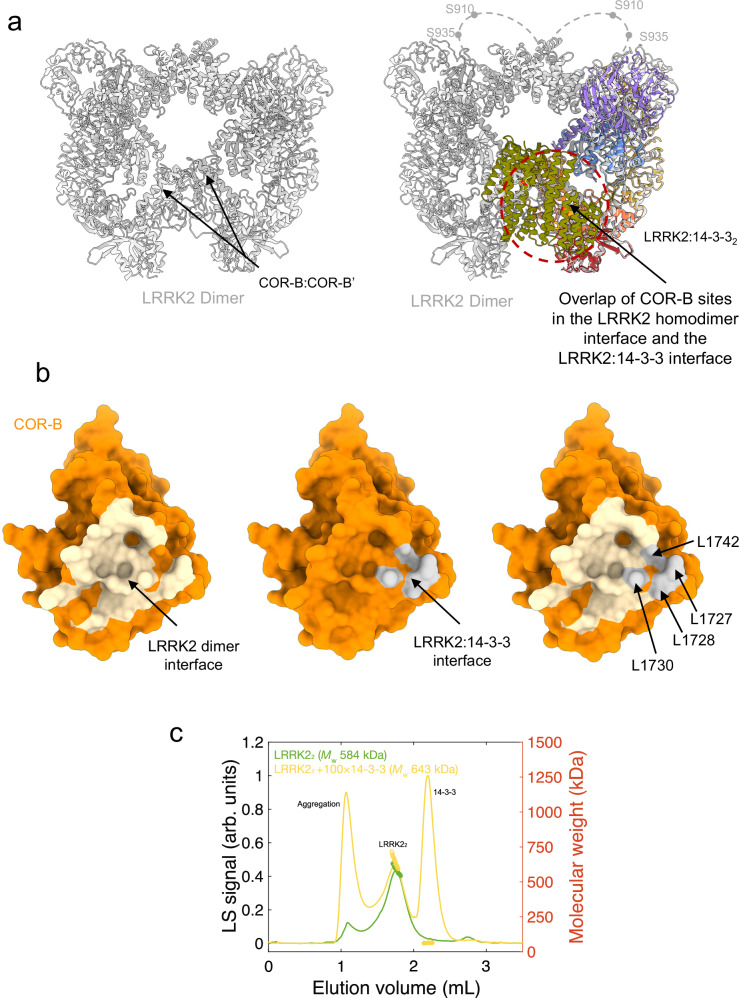
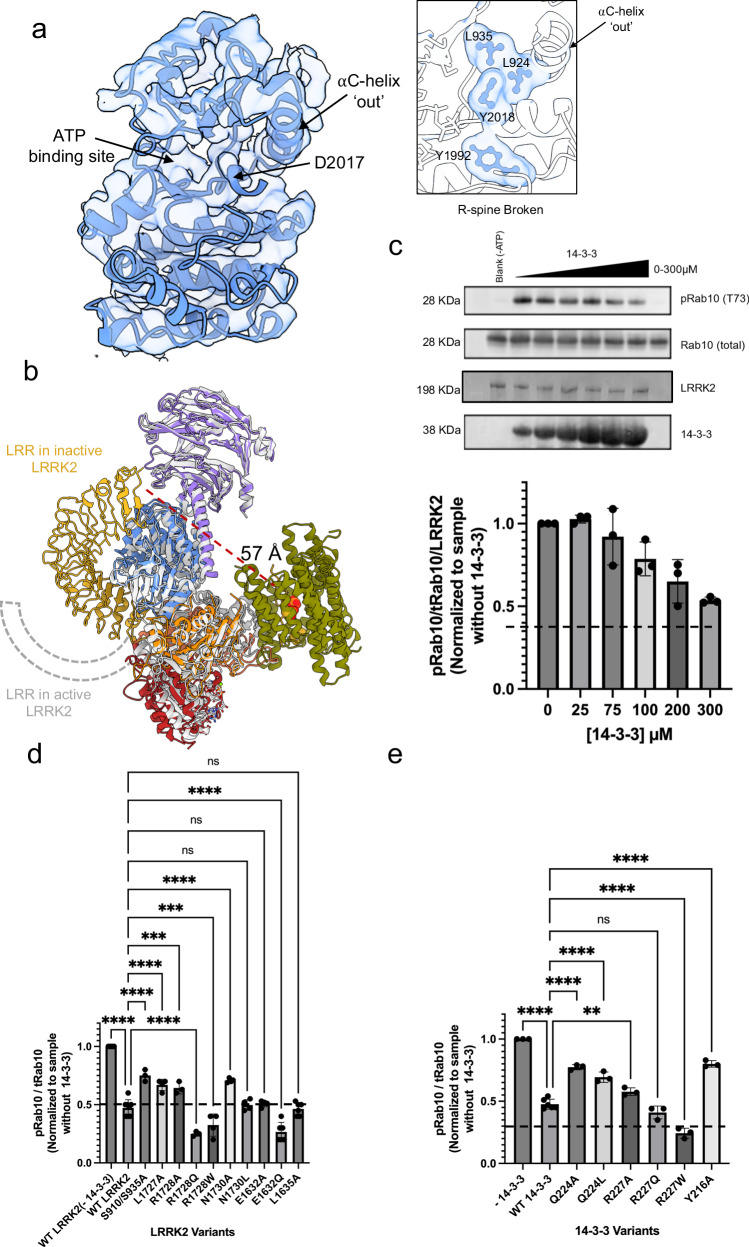
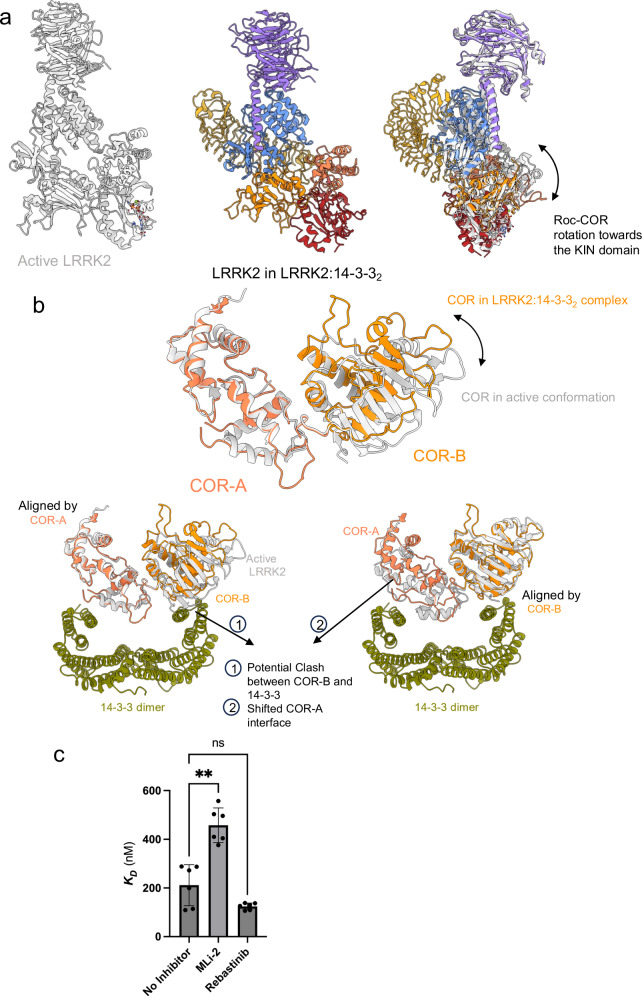
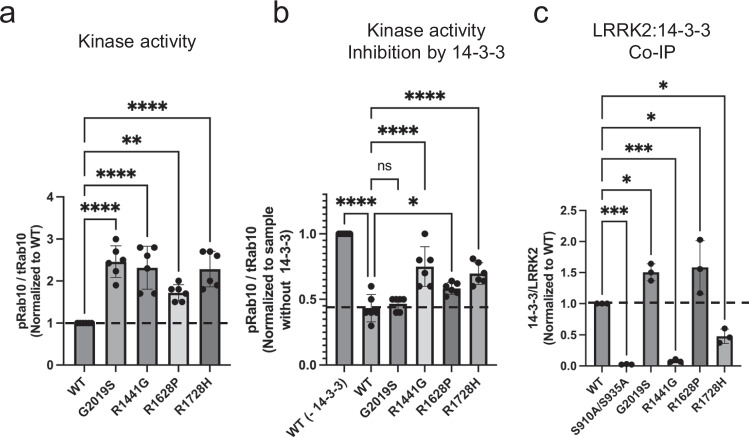
Leucine-rich repeat kinase 2 (LRRK2) is an essential regulator in cellular signaling and a major contributor to Parkinson's disease (PD) pathogenesis. 14-3-3 proteins are critical modulators of LRRK2 activity, yet the structural basis of their interaction has remained unclear. Here, we present the cryo-electron microscopy structure of the LRRK2:14-3-32 autoinhibitory complex, revealing how a 14-3-3 dimer stabilizes an autoinhibited LRRK2 monomer through dual-site anchoring. The dimer engages both phosphorylated S910/S935 sites and the COR-A/B subdomains within the Roc-COR GTPase region. This spatial configuration constrains LRR domain mobility, reinforces the inactive conformation, and likely impedes LRRK2 dimerization and oligomer formation. Structure-guided mutagenesis studies show that PD-associated mutations at the COR:14-3-32 interface and within the GTPase domain weaken 14-3-3 binding and impair its inhibitory effect on LRRK2 kinase activity. Furthermore, we demonstrate that type I LRRK2 kinase inhibitor, which stabilizes the kinase domain in its active conformation, reduces 14-3-3 binding and promotes dephosphorylation at pS910 and pS935. Together, these findings provide a structural basis for understanding how LRRK2 is maintained in an inactive state, elucidate the mechanistic role of 14-3-3 in LRRK2 regulation, inform the interpretation of PD biomarkers, and suggest therapeutic strategies aimed at enhancing LRRK2-14-3-3 interactions to treat PD and related disorders.
© 2025. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Update of
-
14-3-3 binding maintains the Parkinson's associated kinase LRRK2 in an inactive state.bioRxiv [Preprint]. 2024 Nov 22:2024.11.22.624879. doi: 10.1101/2024.11.22.624879. bioRxiv. 2024. Update in: Nat Commun. 2025 Aug 5;16(1):7226. doi: 10.1038/s41467-025-62337-1. PMID: 39605327 Free PMC article. Updated. Preprint.
References
-
- Gasser, T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev. Mol. Med.10.1017/S1462399409001148 (2009). - PubMed
-
- Satake, W. et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet.41, 1303–1307 (2009). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
