

Ivabradine induces RAD51 degradation, potentiating PARP inhibitor efficacy in non-germline BRCA pathogenic variant triple-negative breast cancer
- PMID: 40764992
- PMCID: PMC12323259
- DOI: 10.1186/s12967-025-06902-8
Ivabradine induces RAD51 degradation, potentiating PARP inhibitor efficacy in non-germline BRCA pathogenic variant triple-negative breast cancer
Abstract
Background: Triple-negative breast cancer (TNBC) is an aggressive subtype lacking targetable proteins for treatment. PARP inhibitors (PARPi) are effective in BRCA-mutated cancers but have limited utility in non-germline BRCA-mutated (non-gBRCAm) TNBC. We hypothesized that inducing BRCAness by targeting RAD51, a key homologous recombination protein, could sensitize non-gBRCAm TNBC to PARPi.
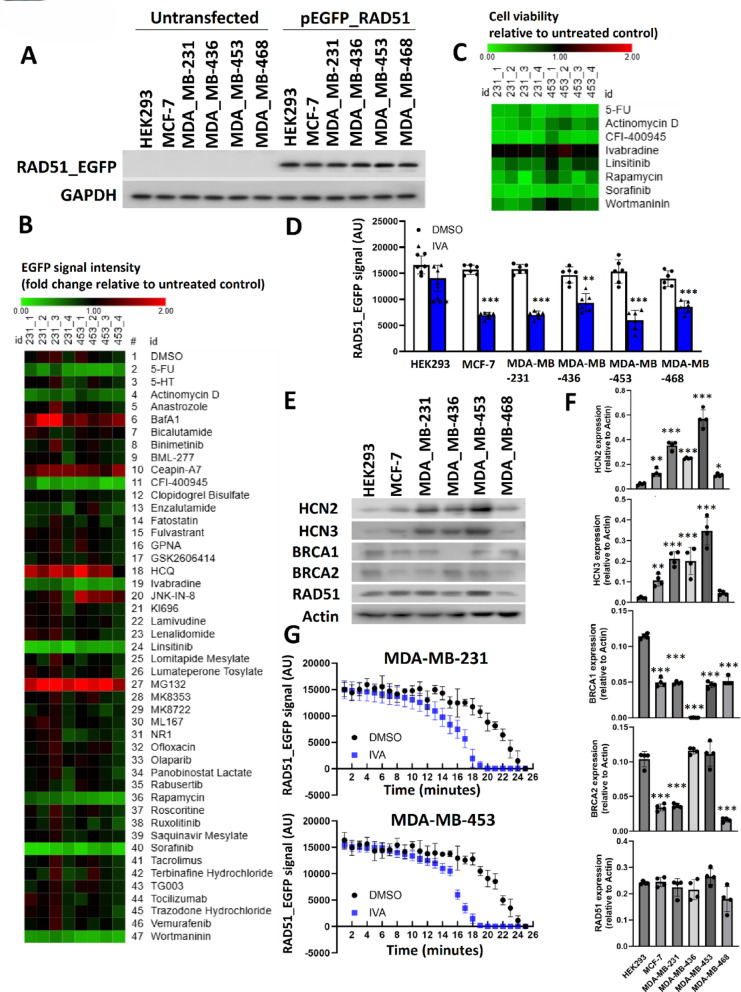
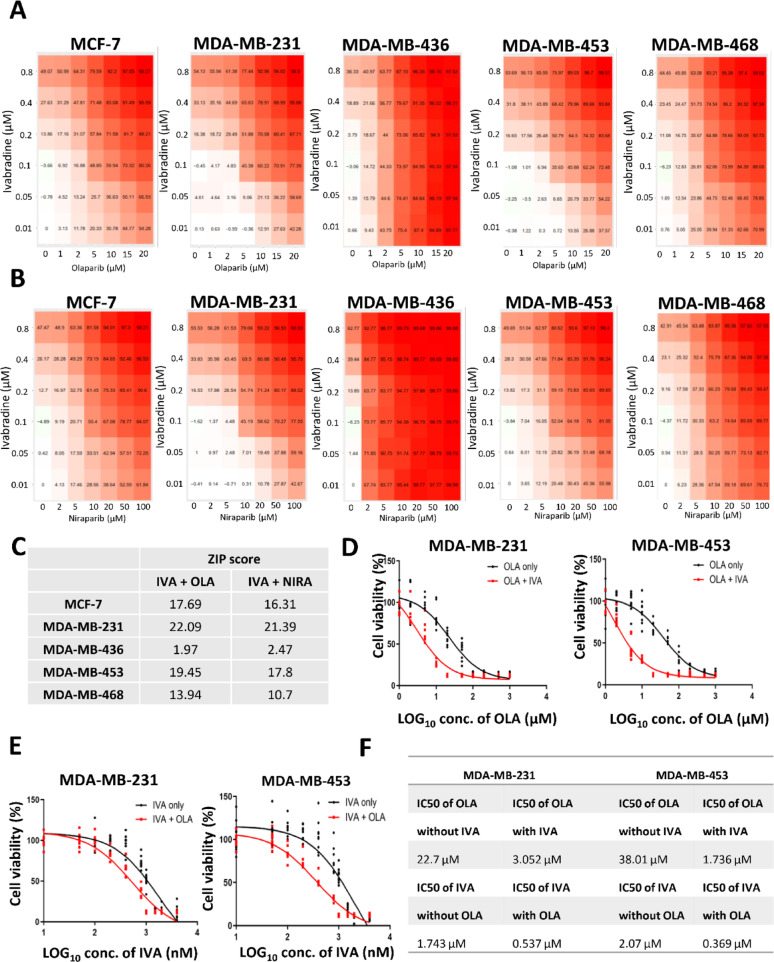
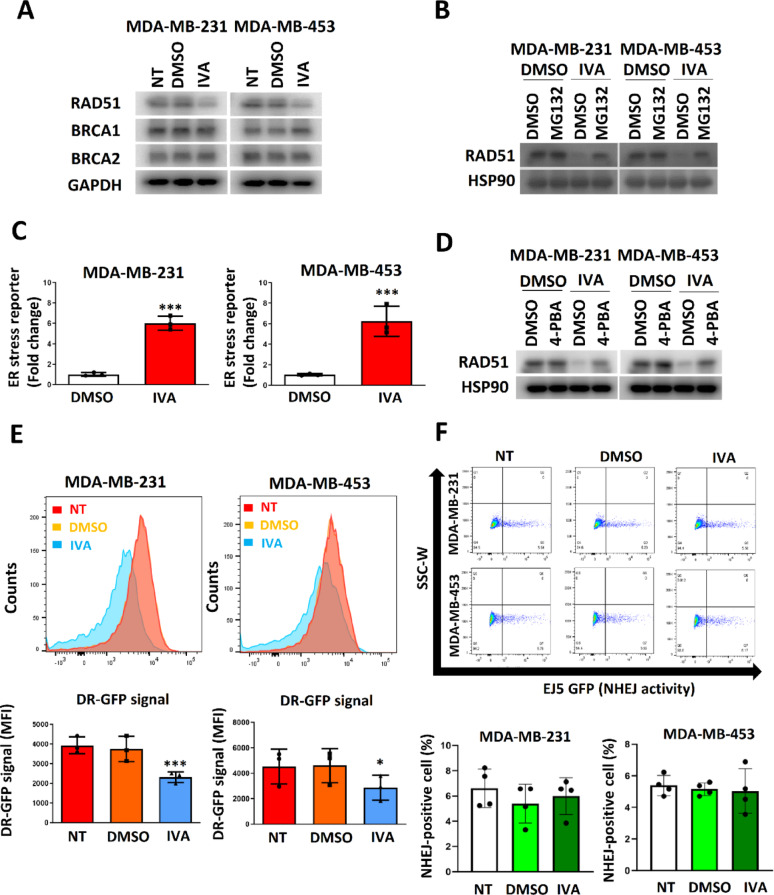
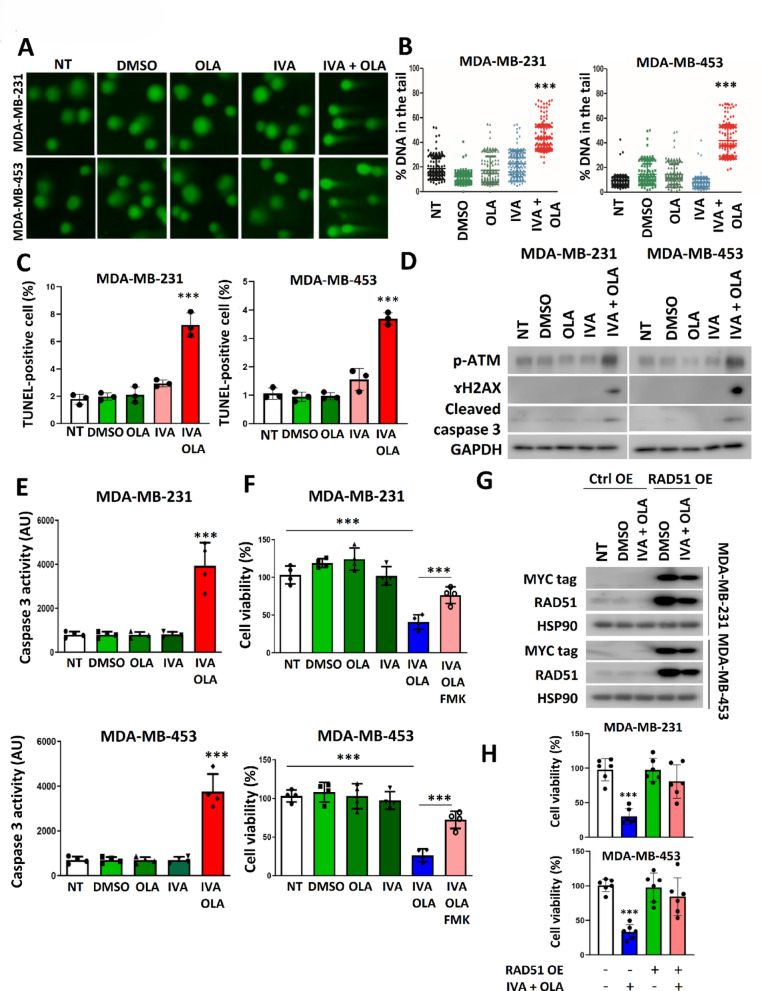
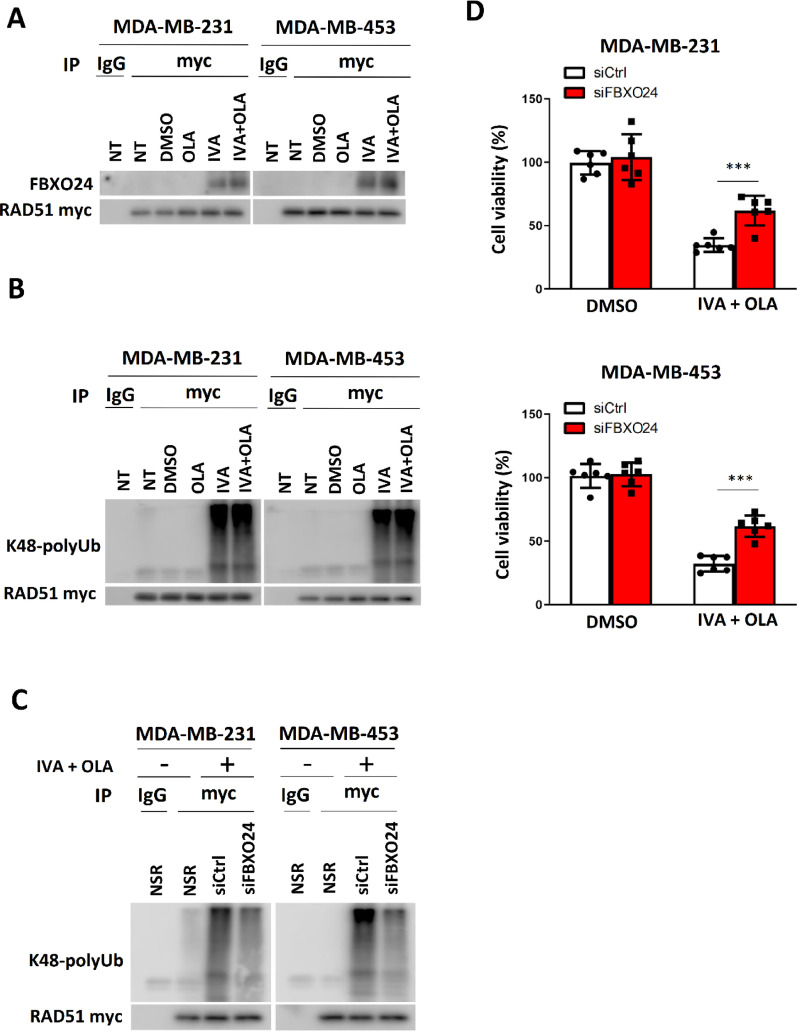
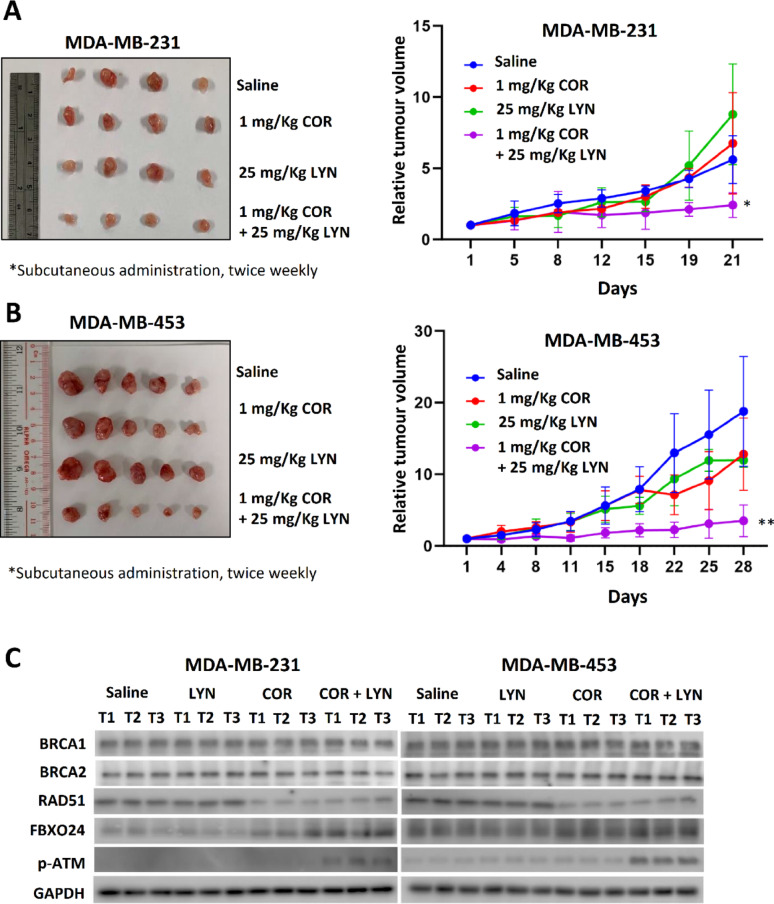
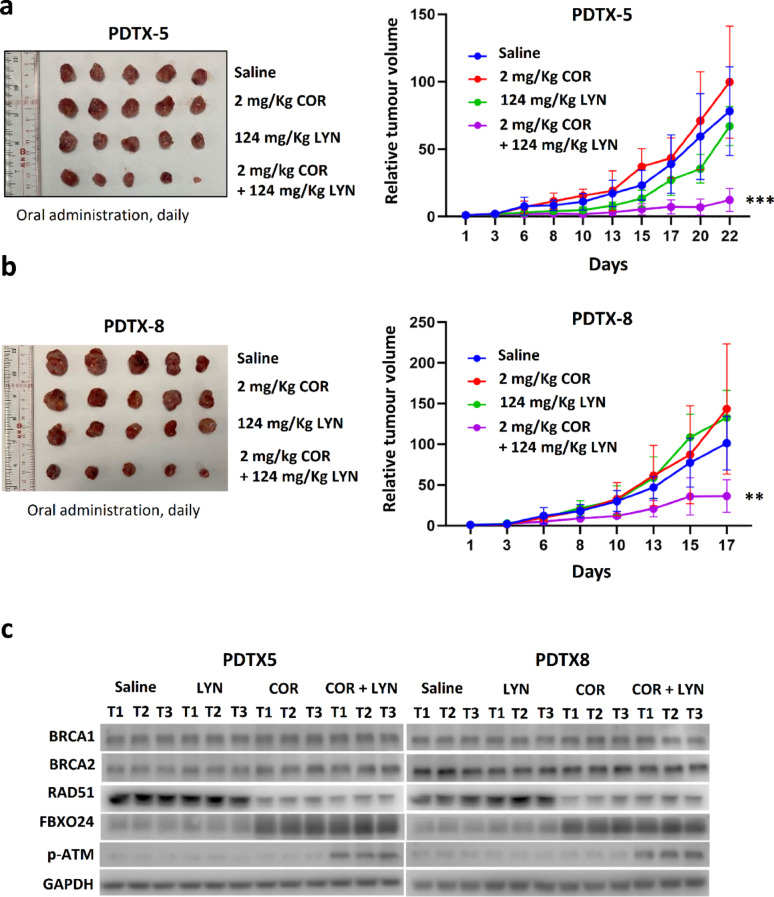
Methods: EGFP-tagged RAD51 was generated and EGFP signal was monitored for identifying agents that affected RAD51 protein expression and stability. Cell viability was assayed using cell counting kit-8. Synergism of ivabradine and olaparib was determined using SynergyFinder 3.0. DR-GFP, EJ5-GFP and comet assays were employed to evaluate the degree of DNA repair and damage, respectively. Protein and mRNA levels were determined by western blot and qPCR, respectively. ChIP was used to determine the binding to ATF6 to the promoter of FBXO24. CoIP was employed to determine the interaction between RAD51 and FBXO24. Xenografts on nude mice and PDTX were in vivo models for validating the combined effect of ivabradine and olaparib.
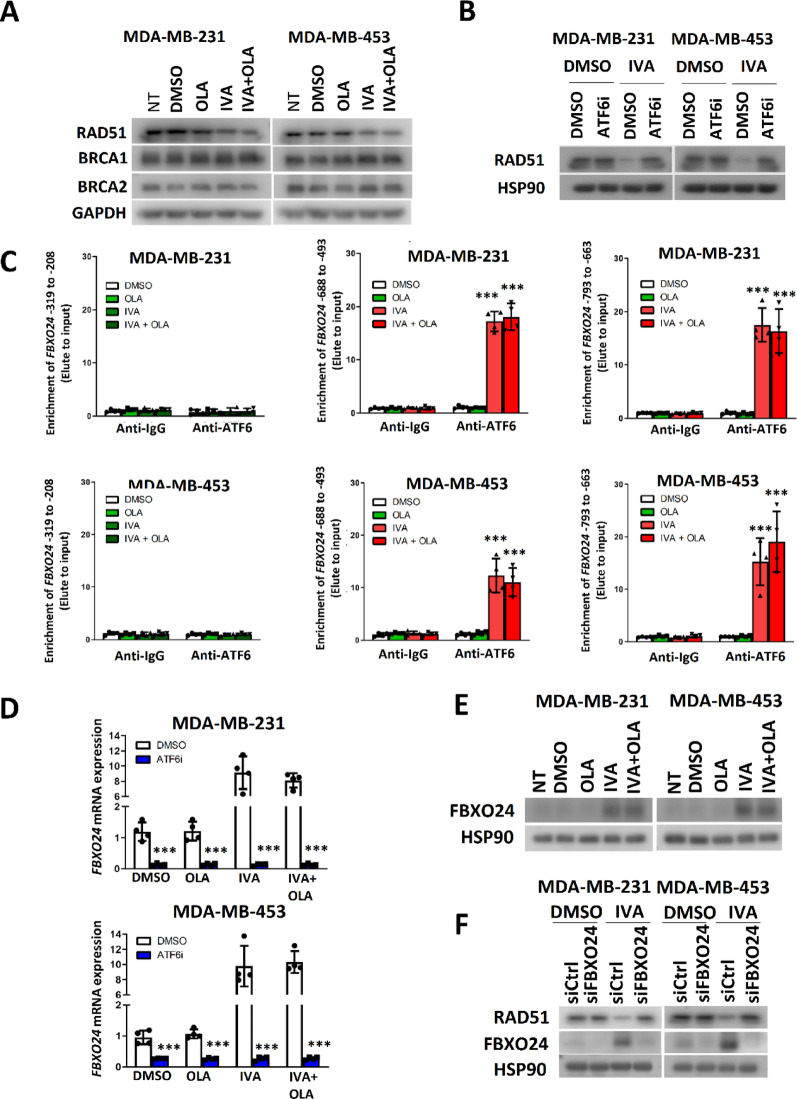
Results: Using an EGFP-RAD51 reporter, we identified ivabradine as a RAD51-reducing agent. In vitro studies with TNBC cell lines demonstrated that ivabradine synergized with PARPi to reduce cell viability (ZIP score > 10), induce apoptosis, and impair HR-mediated DNA repair. This synergy was confirmed in vivo using xenografts and patient-derived tumor xenografts, where co-treatment with clinical grade ivabradine (Coralan) and PARPi olaparib (Lynparza) led to substantial tumor growth inhibition without notable toxicity. Mechanistically, ivabradine triggered ER stress, activating ATF6 to upregulate FBXO24-dependent ubiquitination, leading to RAD51 degradation, resulting in the condition of BRCAness. Chromatin immunoprecipitation and co-immunoprecipitation confirmed the ATF6-FBXO24-RAD51 cascade. These findings reveal a novel mechanism by which ivabradine, an FDA-approved cardiac drug, induces BRCAness, by degrading RAD51 via the ATF6-FBXO24 axis, thus, by mimicking HR deficiency hypersensitizes BRCA-proficient TNBC to olaparib.
Conclusion: This study highlights the translational potential of repurposing ivabradine as a therapeutic strategy for non-gBRCAm TNBC. By addressing a critical unmet need of this aggressive breast cancer subtype, it can potentially expand the utility of PARPi.
Keywords: BRCAness; Breast cancer; Ivabradine; Olaparib; Synthetic lethality; TNBC; Targeted therapy.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: The procedures on mice were reviewed and approved by Committee on the Use of Live Animals in Teaching and Research, The University of Hong Kong (5236-19). Approval was granted from The Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (HKU/HA HKW IRB No. UW 16–391) for collecting human tissue and blood samples. Consent for publication: Not applicable. Competing interests: USK, HT, and CG are the inventors of the patent (Use of HCN Inhibitors for Treatment of Cancer; EP3615052B1). The other authors declare that they have no competing interests.
Figures
References
-
- Schmid P, Cortes J, Dent R, McArthur H, Pusztai L, Kümmel S, Denkert C, Park YH, Hui R, Harbeck N, et al. Overall survival with pembrolizumab in early-stage triple-negative breast cancer. N Engl J Med. 2024;391:1981–91. - PubMed
-
- Burstein HJ. Immunotherapy for early-stage triple-negative breast cancer. N Engl J Med. 2024;391:2048–9. - PubMed
-
- O’Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet. 2017;18:613–23. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
