

This is a preprint.
HNRNPH1-mediated splicing events regulate EIF4G1 transcript variant composition and the organization of the AURKA 5'UTR
- PMID: 40766465
- PMCID: PMC12324280
- DOI: 10.1101/2025.07.28.667222
HNRNPH1-mediated splicing events regulate EIF4G1 transcript variant composition and the organization of the AURKA 5'UTR
Abstract
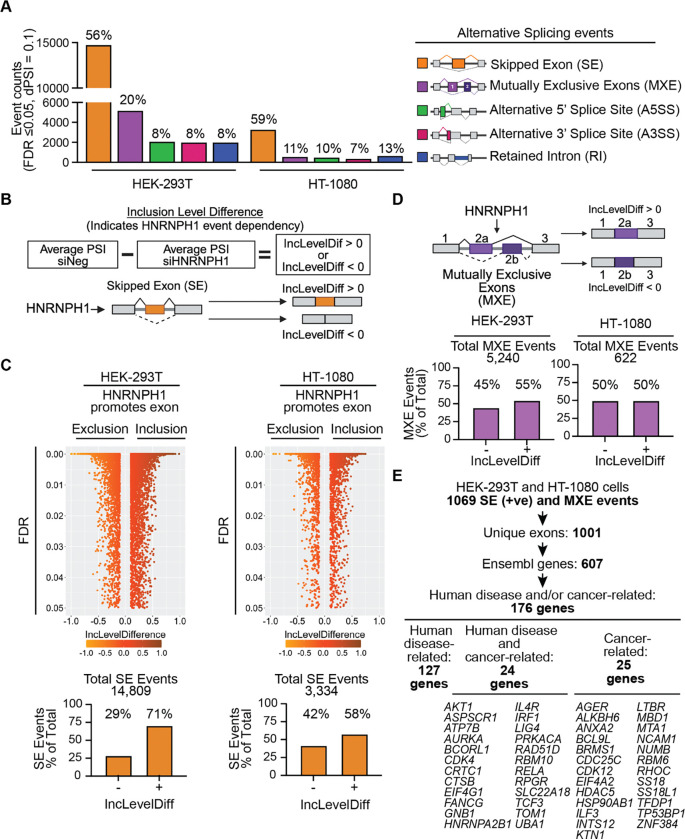
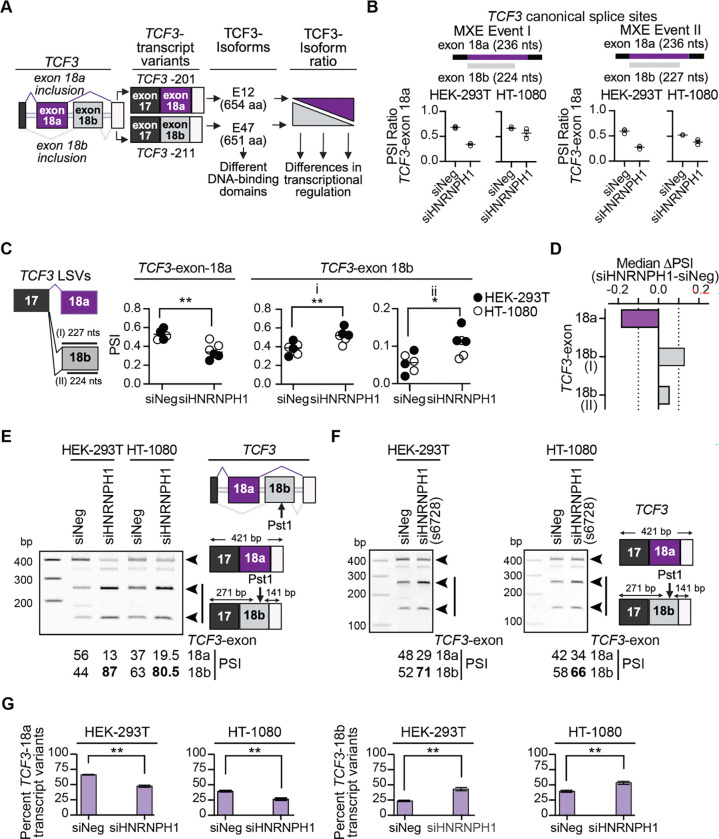
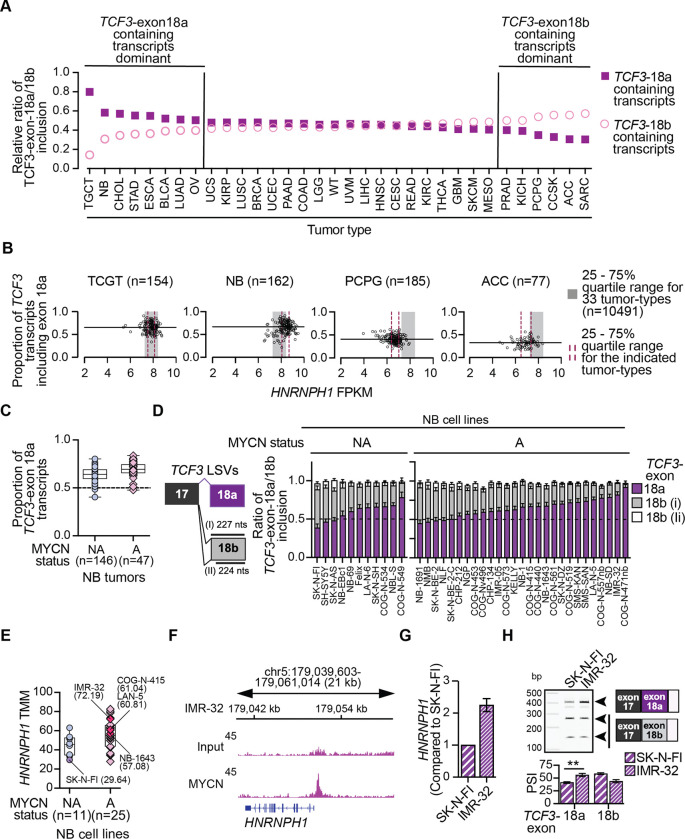
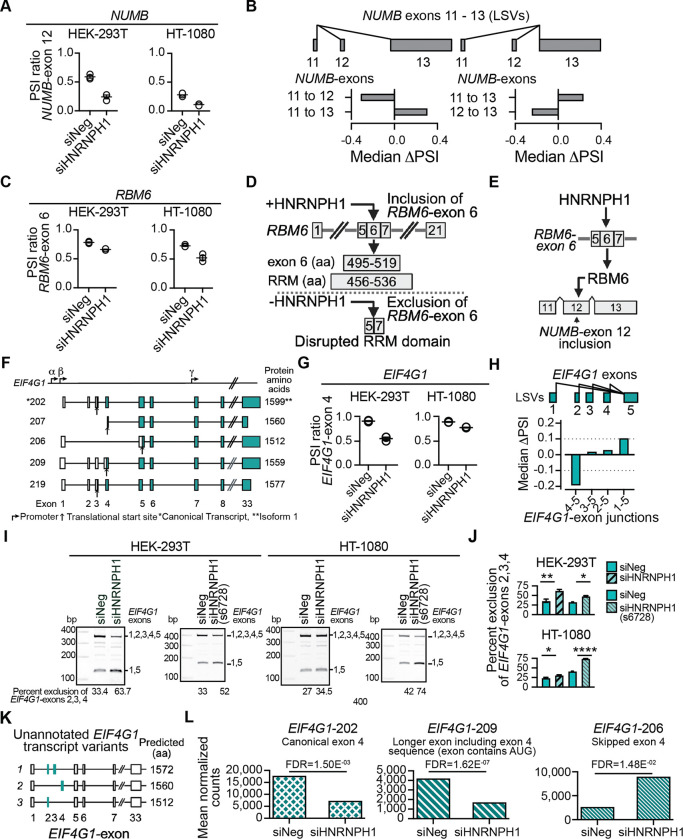
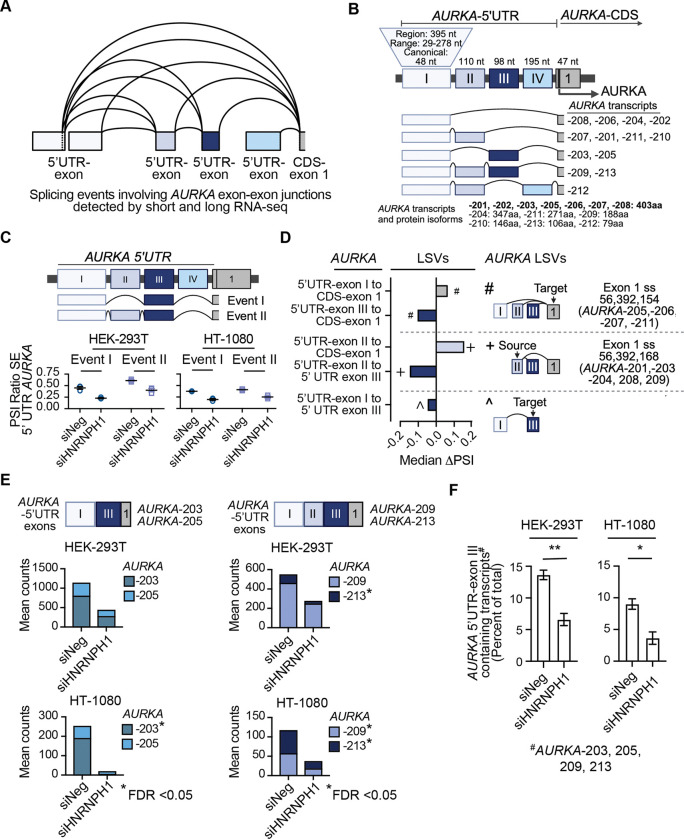
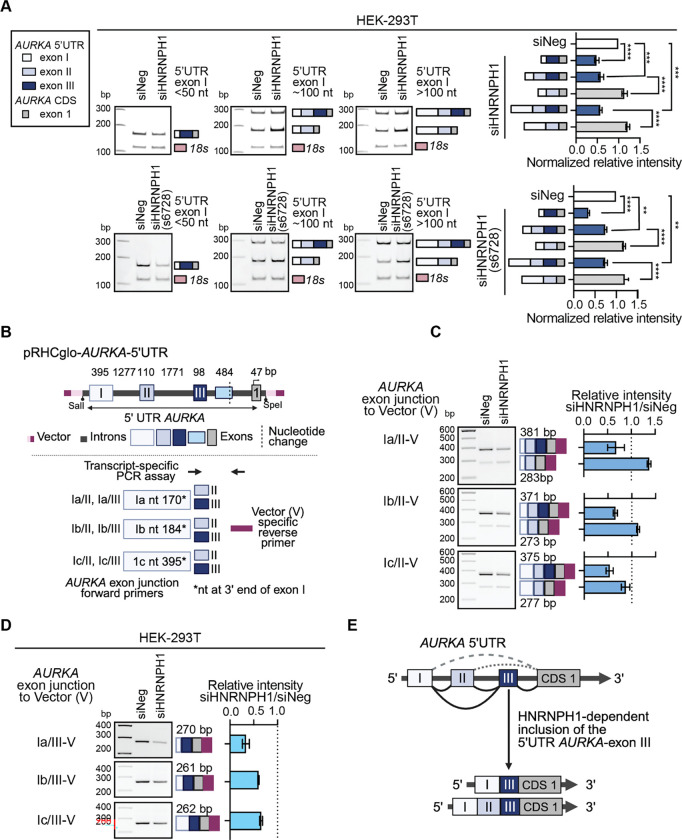
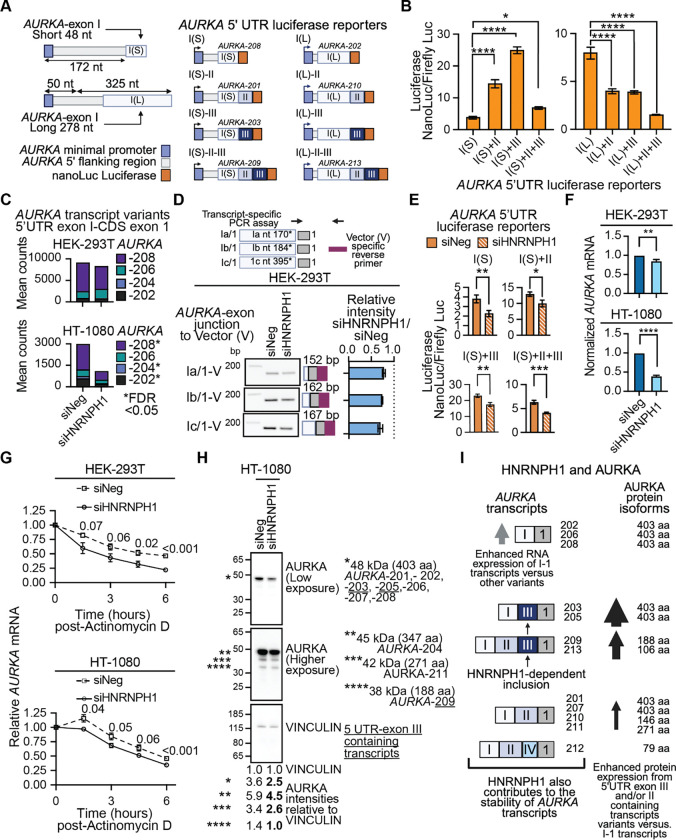
HNRNPH1 is a regulator of alternative splicing, but few studies have defined the splicing events it mediates. Here, we used short- and long-read RNA sequencing to interrogate the transcriptome-wide effects of HNRNPH1 depletion and its regulation of specific splicing events. Differential alternative splicing analysis revealed effects on the transcriptome that involved all splice event categories. We confirmed HNRNPH1's regulation of a splicing event involving TCF3-exons 18a and 18b that encode distinct TCF3 transcription factor isoforms. Extending this finding, we present evidence that in neuroblastoma, HNRNPH1 is a MYCN target, potentially explaining the higher levels of HNRNPH1 and TCF3-exon 18a transcript variants in this tumor type. Analysis of two skipped exon events determined that HNRNPH1 regulates the splicing of exons encoding part of the EIF4G1 translation initiation factor's N-terminus and an exon included in the 5'UTR of specific transcript variants encoding the mitotic kinase AURKA. Using reporter constructs, we show this AURKA 5'UTR exon enhances expression, suggesting HNRNPH1 could contribute to regulating AURKA protein levels. Our findings highlight HNRNPH1's roles in regulating the expression of proteins with diverse cellular functions.
Conflict of interest statement
Conflict of interest statement The authors report no conflicts of interest.
Figures
Similar articles
-
Conservation of alternative splicing in sodium channels reveals evolutionary focus on release from inactivation and structural insights into gating.J Physiol. 2017 Aug 15;595(16):5671-5685. doi: 10.1113/JP274693. Epub 2017 Jul 18. J Physiol. 2017. PMID: 28621020 Free PMC article.
-
SRC kinase isoforms regulate mRNA splicing during neural development.J Neurosci. 2025 Aug 1:e1705242025. doi: 10.1523/JNEUROSCI.1705-24.2025. Online ahead of print. J Neurosci. 2025. PMID: 40750357
-
Positioning of pyrimidine motifs around cassette exons defines their PTB-dependent splicing in Arabidopsis.Plant J. 2024 Jun;118(6):2202-2218. doi: 10.1111/tpj.16739. Epub 2024 Apr 5. Plant J. 2024. PMID: 38578875
-
Antidepressants for pain management in adults with chronic pain: a network meta-analysis.Health Technol Assess. 2024 Oct;28(62):1-155. doi: 10.3310/MKRT2948. Health Technol Assess. 2024. PMID: 39367772 Free PMC article.
-
Survivor, family and professional experiences of psychosocial interventions for sexual abuse and violence: a qualitative evidence synthesis.Cochrane Database Syst Rev. 2022 Oct 4;10(10):CD013648. doi: 10.1002/14651858.CD013648.pub2. Cochrane Database Syst Rev. 2022. PMID: 36194890 Free PMC article.
References
-
- Wilkinson M.E., Charenton C. and Nagai K. (2020) RNA Splicing by the Spliceosome. Annual Review of Biochemistry, Vol 89, 89, 359–388. - PubMed
-
- Pan Q., Shai O., Lee L.J., Frey B.J. and Blencowe B.J. (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet, 40, 1413–1415. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous