

This is a preprint.
A Structure-Based Computational Pipeline for Broad-Spectrum Antiviral Discovery
- PMID: 40766596
- PMCID: PMC12324269
- DOI: 10.1101/2025.07.29.667267
A Structure-Based Computational Pipeline for Broad-Spectrum Antiviral Discovery
Abstract
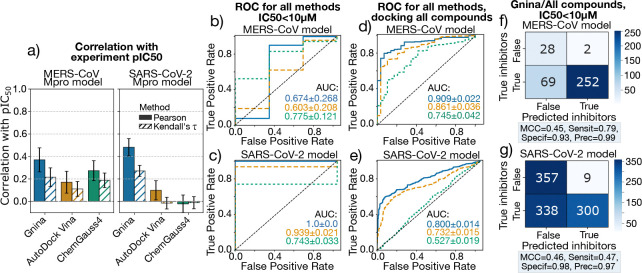
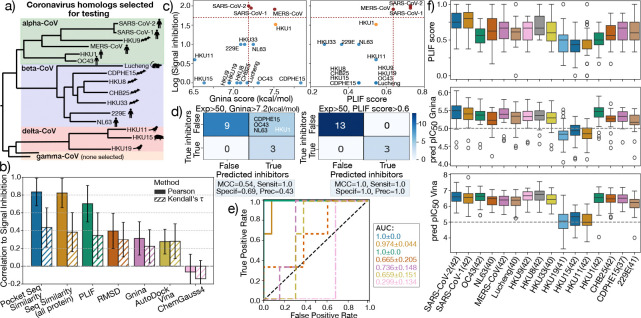
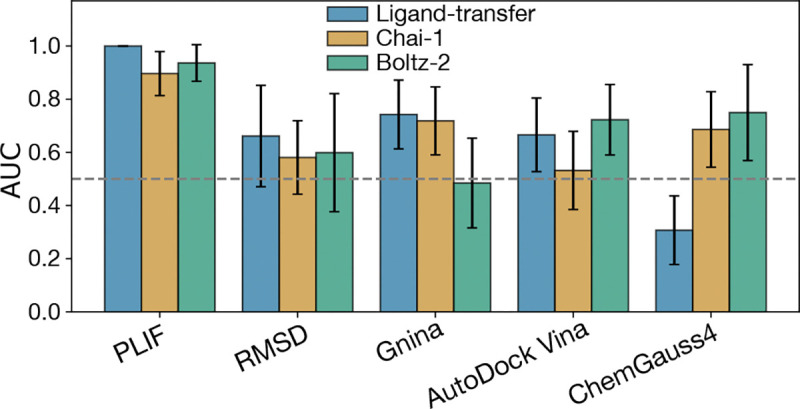
The rapid emergence of viruses with pandemic potential continues to pose a threat to public health worldwide. With the typical drug discovery pipeline taking an average of 5-10 years to reach clinical readiness, there is an urgent need for strategies to develop broad-spectrum antivirals that can target multiple viral family members and variants of concern. We present a structure-based computational pipeline designed to identify and evaluate broad-spectrum inhibitors across viral family members for a given target in order to support spectrum breadth assessment and prioritization in lead optimization programs. This pipeline comprises three key steps: (1) an automated search to identify viral sequences related to a specified target construct, (2) pose prediction leveraging any available structural data, and (3) scoring of protein-ligand complexes to estimate antiviral activity breadth. The pipeline is implemented using the drugforge package: an open-source toolkit for structure-based antiviral discovery. To validate this framework, we retrospectively evaluated two overlapping datasets of ligands bound to the SARS-CoV-2 and MERS-CoV main protease (Mpro), observing useful predictive power with respect to experimental binding affinities. Additionally, we screened known SARS-CoV-2 Mpro inhibitors against a panel of human and non-human coronaviruses, demonstrating the potential of this approach to assess broad-spectrum antiviral activity. Our computational strategy aims to accelerate the identification of antiviral therapies for current and emerging viruses with pandemic potential, contributing to global preparedness for future outbreaks.
Figures
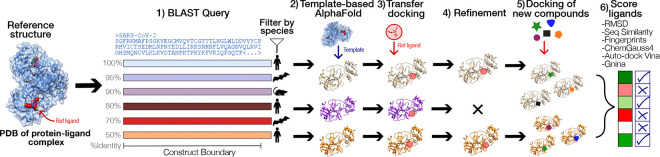
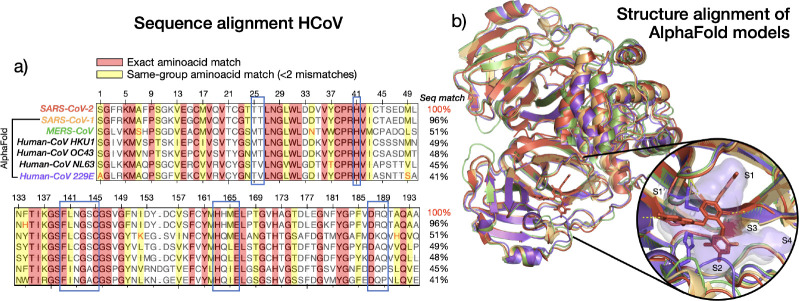
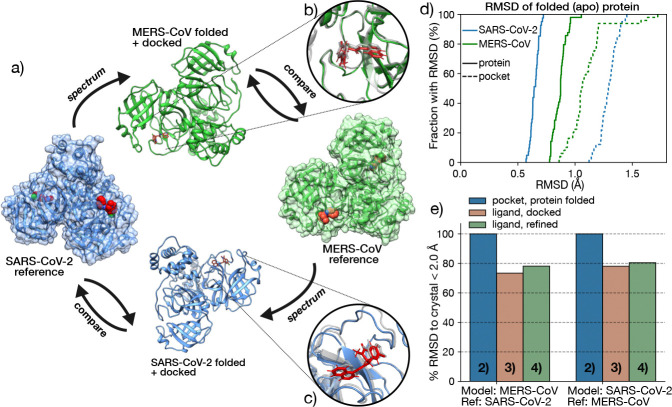
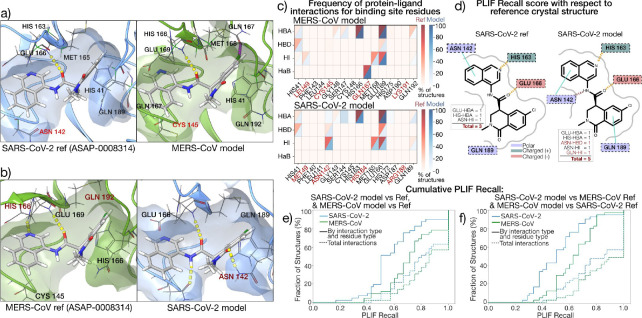
References
-
- PROCEEDINGS OF THE PHYSIOLOGICAL SOCIETY: January 22, 1910. The Journal of Physiology. 1910; 40(suppl):i–vii. https://physoc.onlinelibrary.wiley.com/doi/abs/10.1113/jphysiol.1910.sp0..., doi: 10.1113/jphysiol.1910.sp001386. - DOI - DOI - PMC - PubMed
-
- Ahdritz G, Bouatta N, Floristean C, Kadyan S, Xia Q, Gerecke W, O’Donnell TJ, Berenberg D, Fisk I, Zanichelli N, Zhang B, Nowaczynski A, Wang B, Stepniewska-Dziubinska MM, Zhang S, Ojewole A, Guney ME, Biderman S, Watkins AM, Ra S, et al. OpenFold: Retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. bioRxiv. 2022; https://www.biorxiv.org/content/10.1101/2022.11.20.517210, doi: 10.1101/2022.11.20.517210. - DOI - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous