

This is a preprint.
Discovery of chromatin-based determinants of azacytidine and decitabine anti-cancer activity
- PMID: 40766713
- PMCID: PMC12324281
- DOI: 10.1101/2025.07.29.664810
Discovery of chromatin-based determinants of azacytidine and decitabine anti-cancer activity
Abstract
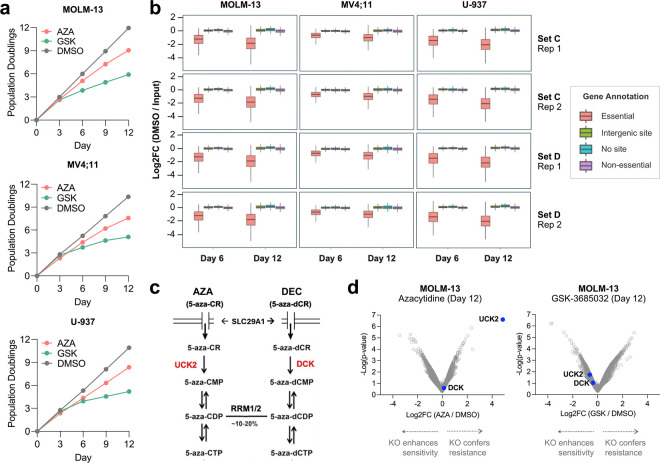
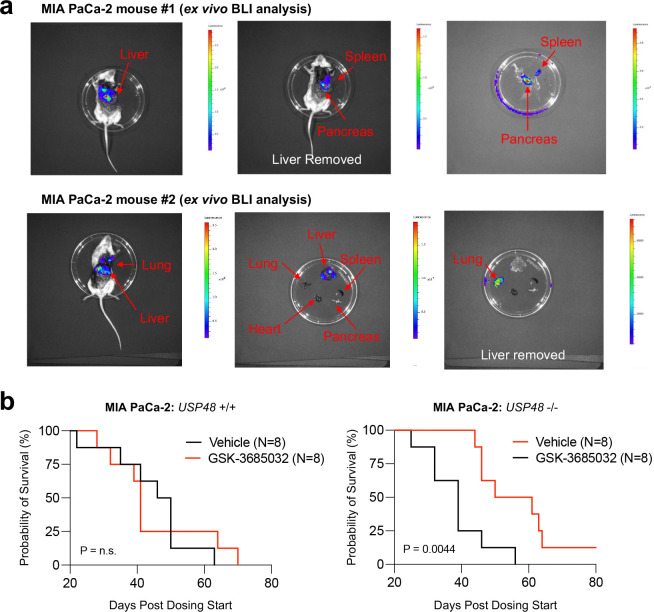
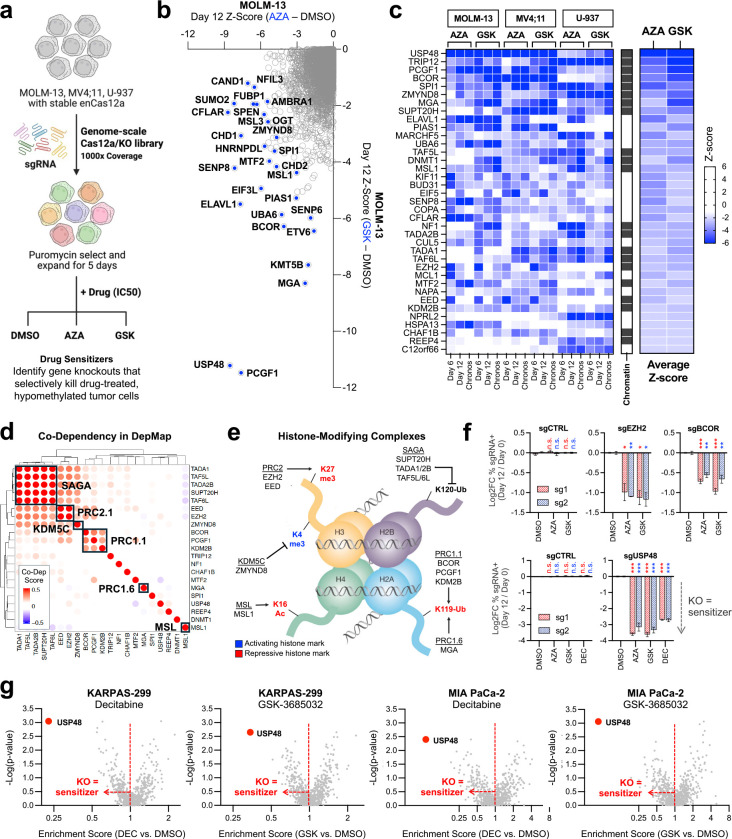
The DNA-incorporating nucleoside analogs azacytidine (AZA) and decitabine (DEC) have clinical efficacy in blood cancers, yet the precise mechanism by which these agents kill cancer cells has remained unresolved - specifically, whether their anti-tumor activity arises from conventional DNA damage or DNA hypomethylation via DNA methyltransferase 1 (DNMT1) inhibition. This incomplete mechanistic understanding has limited their broader therapeutic application, particularly in solid tumors, where early clinical trials showed limited efficacy. Here, through the assessment of drug sensitivity in over 600 human cancer models and comparison to a non-DNA-damaging DNMT1 inhibitor (GSK-3685032), we establish DNA hypomethylation, rather than DNA damage, as the primary killing mechanism of AZA and DEC across diverse cancer types. In further support of an epigenetic killing mechanism, CRISPR drug modifier screens identified a core set of chromatin regulators, most notably the histone deubiquitinase USP48, as AZA and DEC protective factors. We show that USP48 is recruited to newly hypomethylated CpG islands and deubiquitinates non-canonical histones, establishing USP48 as a key molecular link between the two components of epigenetic gene regulation: DNA methylation and chromatin modification. Furthermore, loss of USP48, which occurs naturally through biallelic deletions in human cancers, sensitized both hematologic and solid tumors to DNMT1 inhibition in vitro and in vivo. Our findings elucidate the epigenetic mechanism of action of AZA and DEC and identify a homeostatic link between DNA methylation and chromatin state, revealing new therapeutic opportunities for DNMT1 inhibitors in solid tumors.
Conflict of interest statement
COMPETING INTERESTS STATEMENT R.V.P., M.S., and T.R.G. receive research funding unrelated to this work from Calico Life Sciences LLC. T.R.G. is a paid advisor and/or equity holder in Dewpoint Therapeutics, Sherlock Biosciences, Amplifyer Bio, and Braidwell, Inc.. S.A.C. is a member of the scientific advisory boards of Kymera, PTM BioLabs, MobilION, and PrognomIQ.
Figures







References
-
- Sorm F. & Vesely J. The Activity of a New Antimetabolite, 5-Azacytidine, against Lymphoid Leukaemia in Ak Mice. Neoplasma 11, 123–130 (1964). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials