

Whole-genome sequencing of 490,640 UK Biobank participants
- PMID: 40770095
- PMCID: PMC12443626
- DOI: 10.1038/s41586-025-09272-9
Whole-genome sequencing of 490,640 UK Biobank participants
Abstract
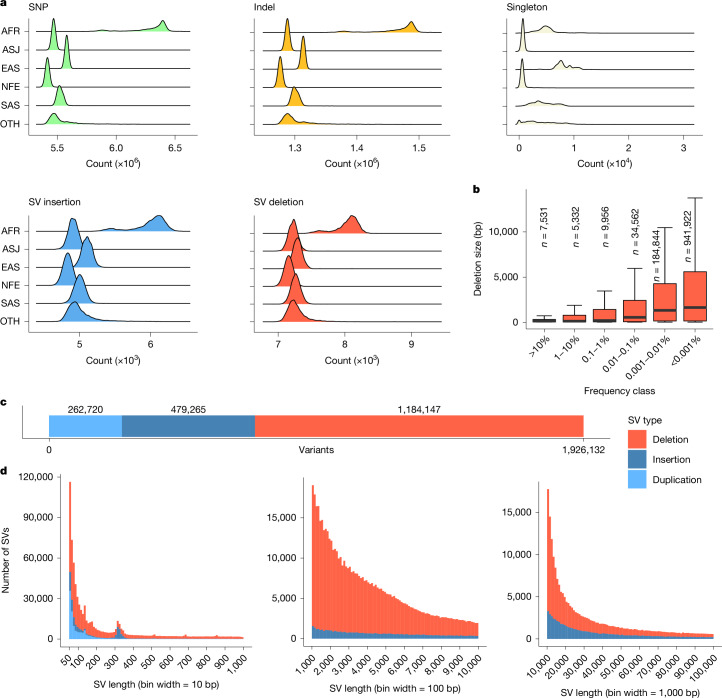
Whole-genome sequencing provides an unbiased and complete view of the human genome and enables the discovery of genetic variation without the technical limitations of other genotyping technologies. Here we report on whole-genome sequencing of 490,640 UK Biobank participants, building on previous genotyping effort1. This advance deepens our understanding of how genetics associates with disease biology and further enhances the value of this open resource for the study of human biology and health. Coupling this dataset with rich phenotypic data, we surveyed within- and cross-ancestry genomic associations and identified novel genetic and clinical insights. Although most associations with disease traits were primarily observed in individuals of European ancestries, strong or novel signals were also identified in individuals of African and Asian ancestries. With the improved ability to accurately genotype structural variants and exonic variation in both coding and UTR sequences, we strengthened and revealed novel insights relative to whole-exome sequencing2,3 analyses. This dataset, representing a large collection of whole-genome sequencing data that is available to the UK Biobank research community, will enable advances of our understanding of the human genome, facilitate the discovery of diagnostics and therapeutics with higher efficacy and improved safety profile, and enable precision medicine strategies with the potential to improve global health.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: K.C., E. Wheeler, K. Kundu, F.H., Q.W., O.S.B., R.S.D., S.V.V.D., C.H., K. Lythgow, P.H.M., K.M., J. Mitchell, S.O., A.O’N., K.R.S., H.T., M.P., R.M., S. Wasilewski and S.P. are or have been employees or contractors of AstraZeneca during the time of this research and may own stock or stock options. J. Liu, Y.L., J. Sandhuria, T.G.R., L. Howe, C.R., D.L., P.A., M.P., D.S., Y.A., J.W., M.D., T.J., J. Davitte, E.I., R.S. and A. Cortes are or have been employees of GSK during the time of this research and may own stock or stock options. B.V.H., H.P.E., K.H.S.M., H. Hauswedell, O.E., A.S., N.G., S. Snorradottir, M.O.U., G.P., M.T.H., A.O., B.O.J., S.K., B.D.S., O.A.S., D. Beyter, G. Holley, V.T., A.G., P.I.O., F.Z., M.A., S.T.S., B. Sigurdsson, S.A.G., G.T.S., G.H.H., G.S., U.S., D.N.M., S.S., K. Kristinsson, E.S., G.T., F.J., P.M., I.J., T.R., H. Holm, H.S., J.S., D.F.G., O.T.M., G.M., U.T., A.H., H.J., P.S. and K.S. are or have been employees of Amgen deCODE genetics and may own stock or stock options. L. Hou, J. Molineros, Y.Z., A.H.L., E.H.B., E.M., A.D.T., G.A.-A., B.M., K.Y.H., J.X., S.N., A.K., S.X., B.F., T.M., T.H. and S.L. are employees and/or stockholders of Janssen Research & Development. R.M., Z.H. and O.S.-T. are employees and/or stockholders of Illumina. The other authors declare no competing interests.
Figures







References
-
- Van Hout, C. V. et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature586, 749–756 (2020).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
