

Genomic analysis of the 2017 Aotearoa New Zealand outbreak of Mycoplasma bovis and its position within the global population structure
- PMID: 40771686
- PMCID: PMC12326744
- DOI: 10.3389/fmicb.2025.1600146
Genomic analysis of the 2017 Aotearoa New Zealand outbreak of Mycoplasma bovis and its position within the global population structure
Abstract
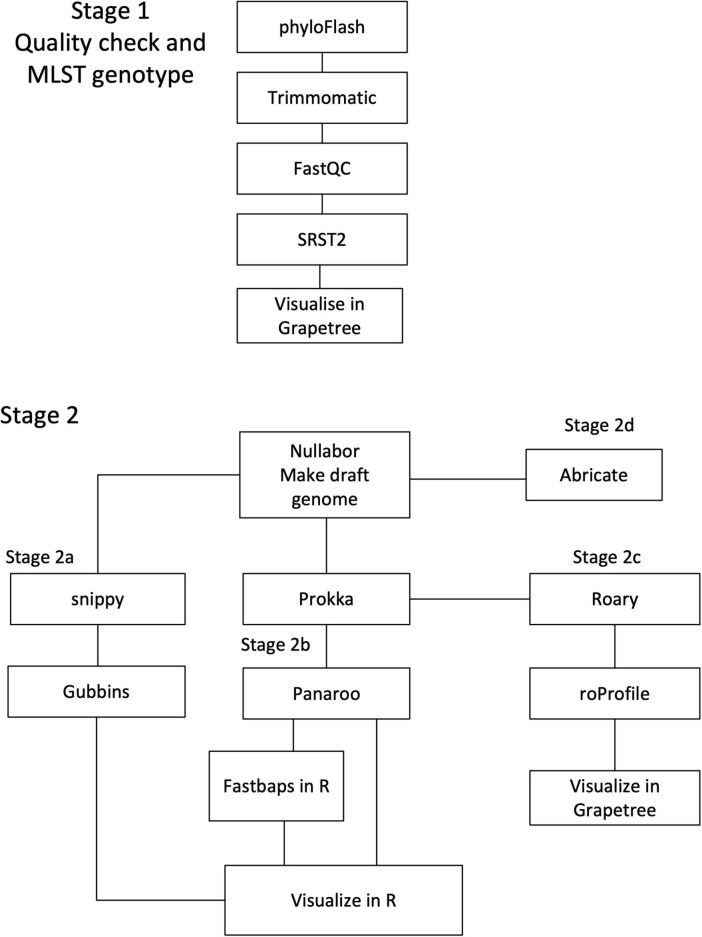
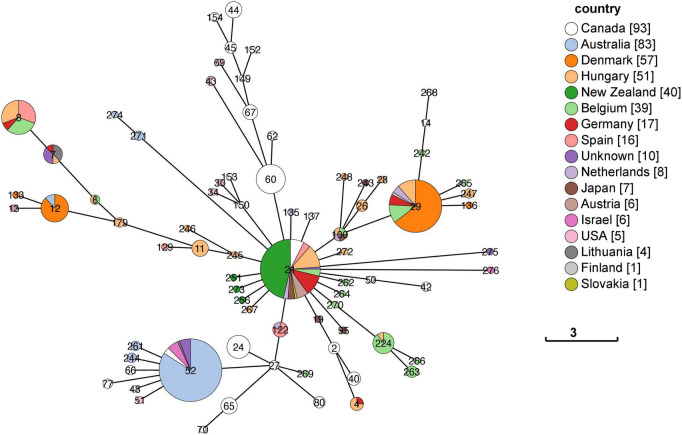
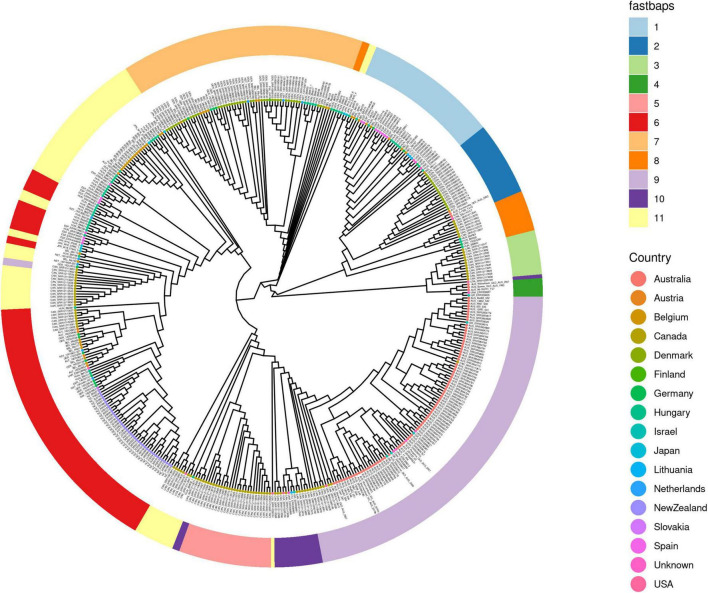
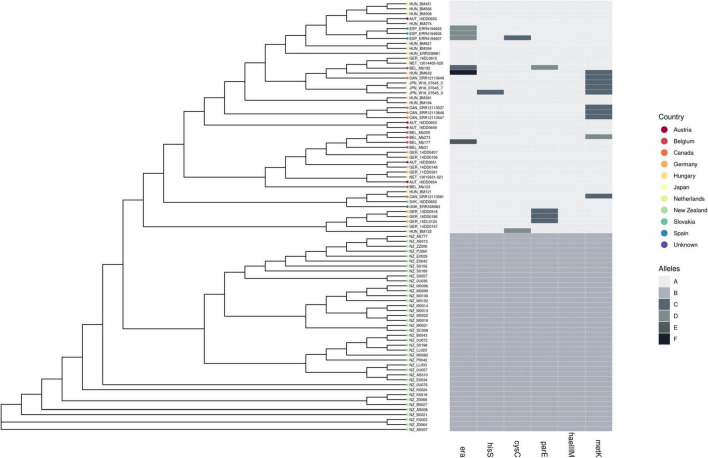
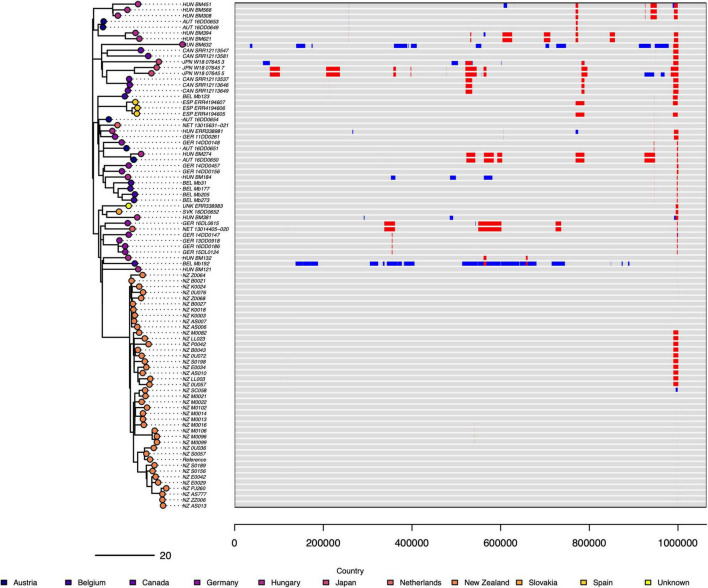
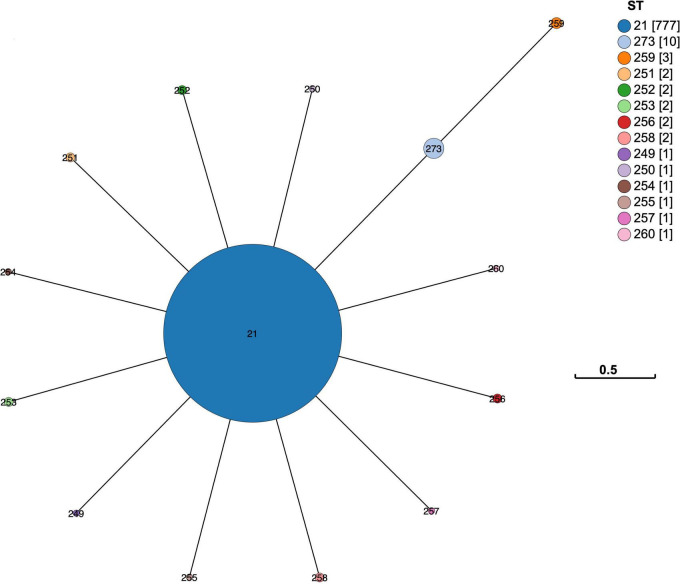
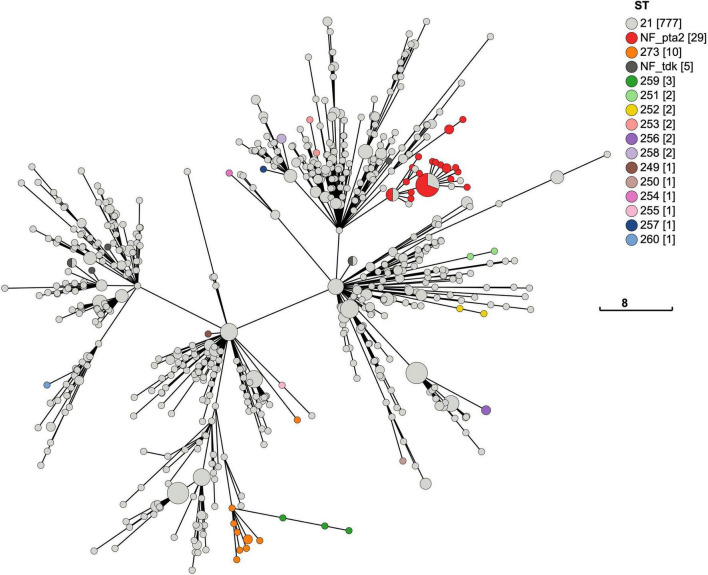
In 2017 an outbreak of Mycoplasma bovis (M. bovis), an infectious agent of cattle, was identified in Aotearoa New Zealand. This study characterizes the genomic population structure of the outbreak in New Zealand and compares it with the known global population structure using multilocus sequence typing (MLST) and genomic analysis. The New Zealand outbreak strain was MLST genotyped as ST21. A comprehensive collection of 840 genomes from the New Zealand outbreak showed a pattern of clonal expansion when characterized by MLST, core genome MLST (cgMLST) and whole genome MLST (wgMLST). A lineage of genomes was found with no in silico identifiable pta2 locus, a housekeeping gene used in the MLST scheme. We compared a sample set of 40 New Zealand genomes to 47 genomes from other countries. This group had 79 ST21 genomes and eight genomes that were single nucleotide polymorphism (SNP) variants within the MLST loci of ST21. Two of the 47 international genomes showed signs of extensive unique recombination. Unique alleles in six genes were identified as present only in the New Zealand genomes. These novel variants were in the genes; haeIIIM encoding for cytosine-specific methyltransferase, cysC encoding for cysteinyl tRNA synthetase, era encoding for GTPase Era, metK encoding for S-adenosylmethionine synthase, parE encoding for DNA topoisomerase, and hisS encoding for histidine-tRNA ligase. This finding could be due to a population bottleneck, genetic drift, or positive selection. The same sample set of 40 New Zealand genomes were compared using MLST to 404 genomes from 15 other countries and 11 genomes without a known country. A FastBAPS analysis of 455 genomes showed a global population structure with 11 clusters. Some countries, such as Canada, Denmark and Australia contained both internally closely related genomes and some genomes that were more closely related to genomes found in other countries. Our results support the need for Whole Genome Sequencing (WGS) as well as MLST genotyping in M. bovis outbreaks. They also support the importance of understanding the national and international movement patterns of cattle and their genetic material, as possible routes of transmission, when managing the spread of M. bovis.
Keywords: Mycoplasma bovis; core genome MLST; genomic epidemiology; multilocus sequence typing (MLST); outbreak; whole genome MLST.
Copyright © 2025 Binney, Gias, Foxwell, Little, Biggs, French, Lambert, Ha, Carter, Gyuranecz, Pardon, De Vliegher, Boyen, Bokma, Krömker, Wente, Mahony, Gibson, Barnes, Wawegama, Legione, Heller, Schnee, Pelkonen, Autio, Higuchi, Gondaira and McCulley.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
-
Diagnostic test accuracy and cost-effectiveness of tests for codeletion of chromosomal arms 1p and 19q in people with glioma.Cochrane Database Syst Rev. 2022 Mar 2;3(3):CD013387. doi: 10.1002/14651858.CD013387.pub2. Cochrane Database Syst Rev. 2022. PMID: 35233774 Free PMC article.
-
Can a Liquid Biopsy Detect Circulating Tumor DNA With Low-passage Whole-genome Sequencing in Patients With a Sarcoma? A Pilot Evaluation.Clin Orthop Relat Res. 2025 Jan 1;483(1):39-48. doi: 10.1097/CORR.0000000000003161. Epub 2024 Jun 21. Clin Orthop Relat Res. 2025. PMID: 38905450
-
Sexual Harassment and Prevention Training.2024 Mar 29. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Mar 29. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 36508513 Free Books & Documents.
-
Factors that influence participation in physical activity for people with bipolar disorder: a synthesis of qualitative evidence.Cochrane Database Syst Rev. 2024 Jun 4;6(6):CD013557. doi: 10.1002/14651858.CD013557.pub2. Cochrane Database Syst Rev. 2024. PMID: 38837220 Free PMC article. Review.
References
-
- Andrews S. (2010). FastQC: A quality control tool for high throughput sequence data. Cambridge: Babraham Bioinformata.
LinkOut - more resources
Full Text Sources