

Dravet syndrome: novel insights into SCN1A-mediated epileptic neurodevelopmental disorders within the molecular diagnostic-therapeutic framework
- PMID: 40772259
- PMCID: PMC12326748
- DOI: 10.3389/fnins.2025.1634718
Dravet syndrome: novel insights into SCN1A-mediated epileptic neurodevelopmental disorders within the molecular diagnostic-therapeutic framework
Abstract
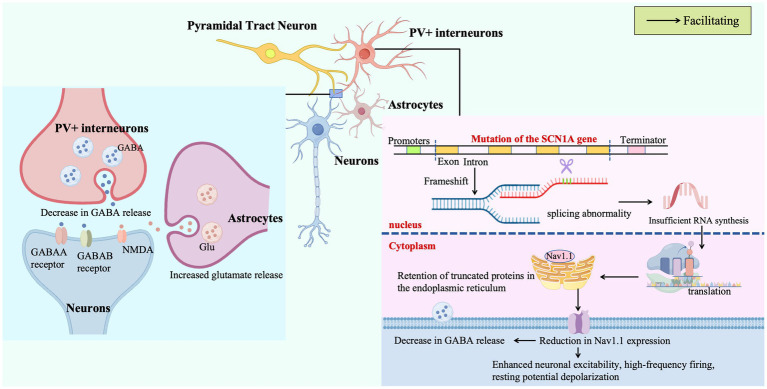
Dravet Syndrome (DS), a rare genetic encephalopathy characterized by severe drug-resistant epilepsy and progressive neurodevelopmental regression in infancy, is caused by de novo mutations in the SCN1A gene on chromosome 2q24 in over 80% of cases. This review synthesizes current insights into its molecular pathogenesis, precision diagnostics, and therapeutic innovations: SCN1A mutations disrupt Nav1.1 sodium channel expression and membrane trafficking in GABAergic interneurons through transcriptional dysregulation, pre-mRNA splicing defects, and gating dysfunction, thereby impairing inhibitory synaptic transmission and disrupting brainwide excitatory-inhibitory balance. Notably, polygenic interactions (e.g., DEPDC5, CHD2 variants), astrocytic calcium signaling aberrations, and mitochondrial metabolic deficits synergistically exacerbate network hyperexcitability. Diagnostic advancements include a stratified framework integrating early febrile seizure phenotypes, comprehensive SCN1A sequencing (including deep intronic variants), and multimodal assessments (e.g., γ-band EEG power analysis and hippocampal volumetry), which significantly accelerate clinical diagnosis and reduce misdiagnosis. Therapeutic strategies are evolving from empirical seizure control to mechanism-targeted interventions: antisense oligonucleotides (ASOs) restore SCN1A transcript integrity by blocking pathogenic exon inclusion; adeno-associated virus (AAV9)-mediated activation of GABAergic neuron-specific SCN1A promoters and CRISPR/dCas9-driven endogenous Nav1.1 upregulation have both been shown to improve inhibitory synaptic function and elevate seizure thresholds in preclinical models. Additionally, novel molecules such as the Nav1.1-selective agonist Hm1a and 5HT2BR receptor modulators offer new avenues by remodeling neuronal electrophysiology and neurotransmitter homeostasis. By dissecting the multi-dimensional molecular networks underlying DS and highlighting interdisciplinary integration of diagnostic-therapeutic technologies, this review provides a theoretical foundation for developing SCN1A-centric precision medicine, advocating a shift from symptomatic management to mechanism-driven interventions in clinical practice.
Keywords: Dravet syndrom; GABAergic; SCN1A; antisence oligonucleotides; molecular.
Copyright © 2025 Zhang, Huang, Wei, Wu, Xie and Wang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures



Similar articles
-
Antisense oligonucleotides restore excitability, GABA signalling and sodium current density in a Dravet syndrome model.Brain. 2024 Apr 4;147(4):1231-1246. doi: 10.1093/brain/awad349. Brain. 2024. PMID: 37812817 Free PMC article.
-
Ion channels and G protein-coupled receptors: Cannabidiol actions on disorders of excitability and synaptic excitatory-inhibitory ratio.Mol Pharmacol. 2025 Mar;107(3):100017. doi: 10.1016/j.molpha.2025.100017. Epub 2025 Feb 7. Mol Pharmacol. 2025. PMID: 40048808 Review.
-
Interneuron-specific dual-AAV SCN1A gene replacement corrects epileptic phenotypes in mouse models of Dravet syndrome.Sci Transl Med. 2025 Mar 19;17(790):eadn5603. doi: 10.1126/scitranslmed.adn5603. Epub 2025 Mar 19. Sci Transl Med. 2025. PMID: 40106582
-
Neurodevelopmental defects in Dravet syndrome Scn1a+/- mice: Targeting GABA-switch rescues behavioral dysfunctions but not seizures and mortality.Neurobiol Dis. 2025 Apr;207:106853. doi: 10.1016/j.nbd.2025.106853. Epub 2025 Feb 26. Neurobiol Dis. 2025. PMID: 40021096
-
Management of urinary stones by experts in stone disease (ESD 2025).Arch Ital Urol Androl. 2025 Jun 30;97(2):14085. doi: 10.4081/aiua.2025.14085. Epub 2025 Jun 30. Arch Ital Urol Androl. 2025. PMID: 40583613 Review.
References
-
- Alonso C., García-Culebras A., Satta V., Hernández-Fisac I., Sierra Á., Guimaré J. A., et al. (2025). Investigation in blood-brain barrier integrity and susceptibility to immune cell penetration in a mouse model of Dravet syndrome. Brain Behav. Immun. Health 44:100955. doi: 10.1016/j.bbih.2025.100955, PMID: - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources