

Integrative multi-omics reveals a regulatory and exhausted T-cell landscape in CLL and identifies galectin-9 as an immunotherapy target
- PMID: 40775219
- PMCID: PMC12331977
- DOI: 10.1038/s41467-025-61822-x
Integrative multi-omics reveals a regulatory and exhausted T-cell landscape in CLL and identifies galectin-9 as an immunotherapy target
Abstract
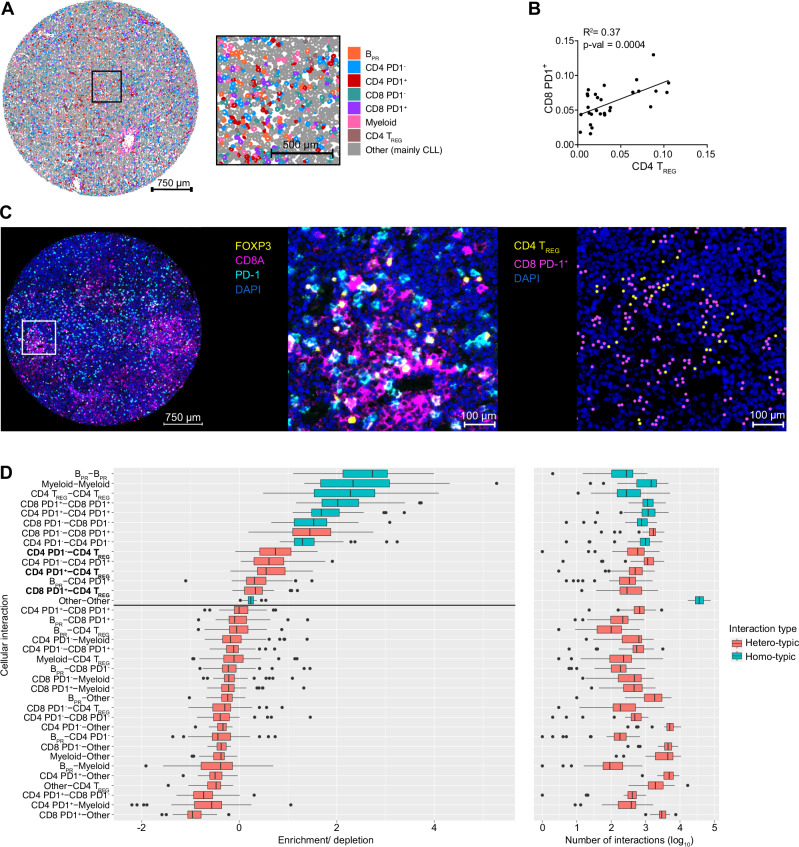
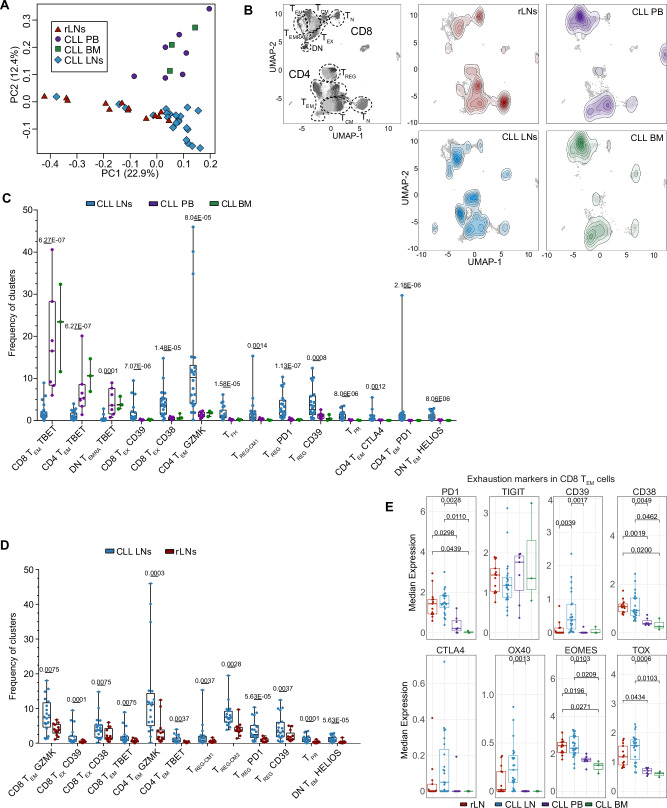
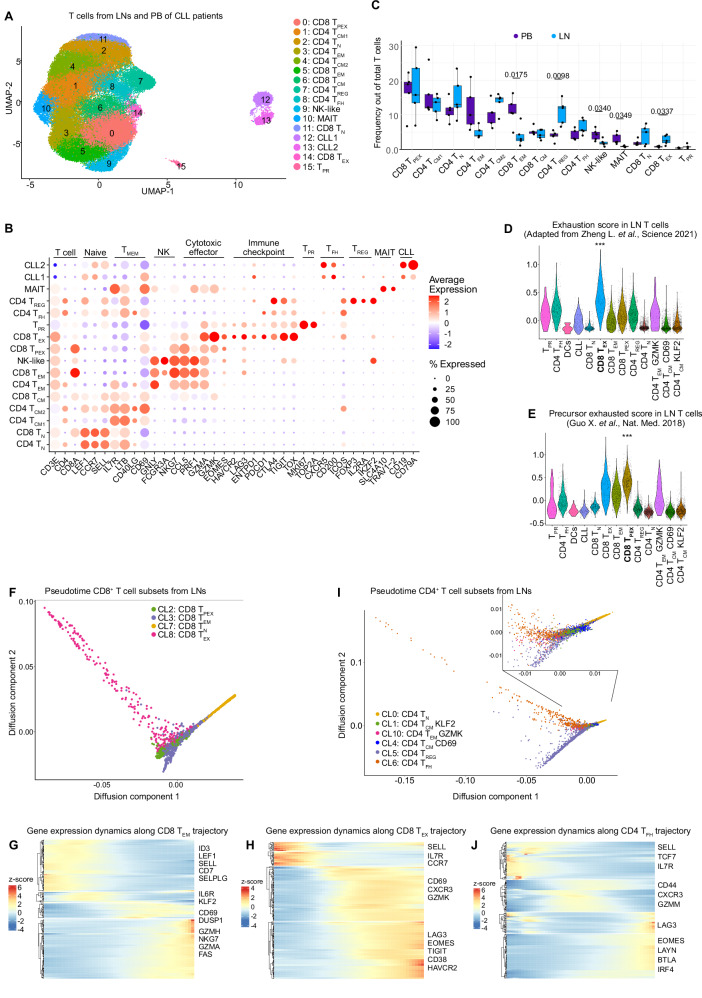
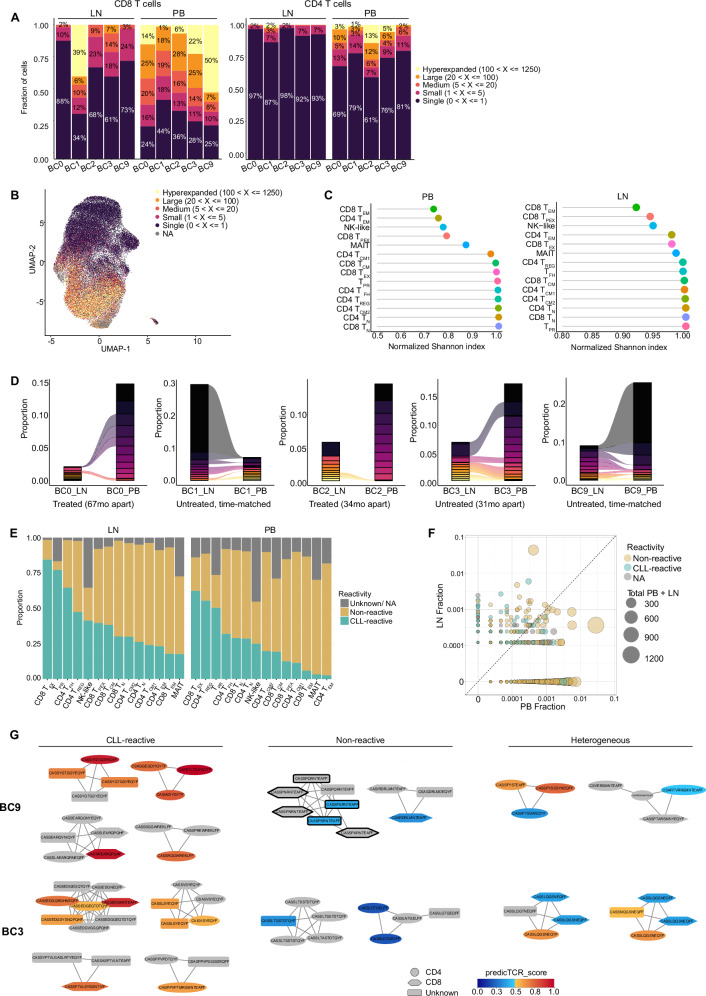
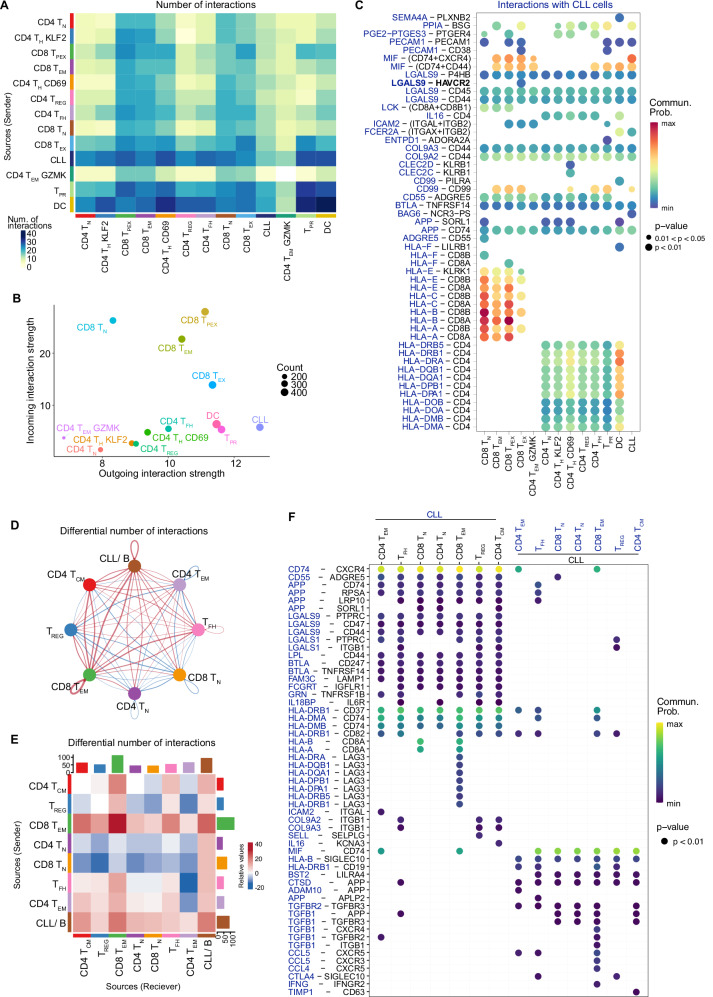
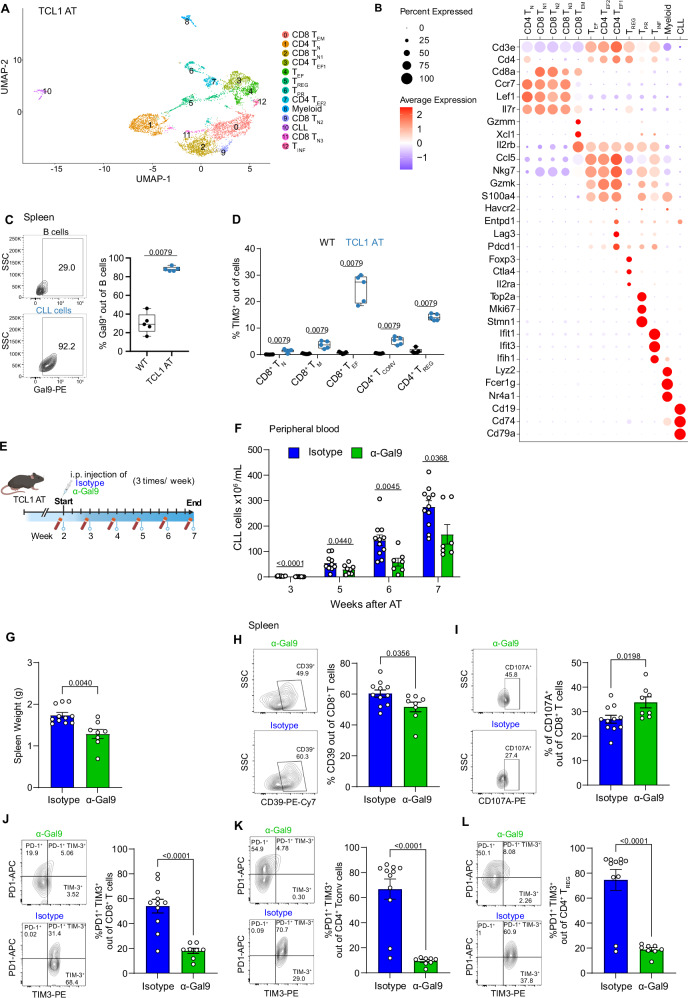
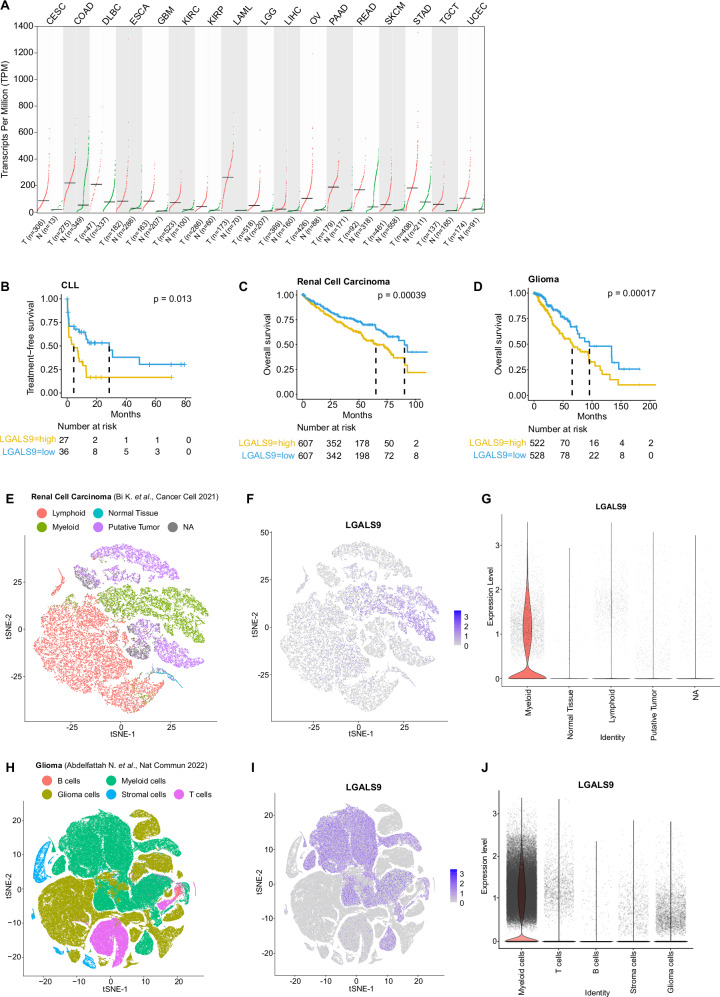
T-cell exhaustion contributes to immunotherapy failure in chronic lymphocytic leukemia (CLL). Here, we analyze T cells from CLL patients' blood, bone marrow, and lymph nodes, as well as from a CLL mouse model, using single-cell RNA sequencing, mass cytometry, and tissue imaging. T cells in CLL lymph nodes show the most distinct profiles, with accumulation of regulatory T cells and CD8+ T cells in various exhaustion states, including precursor (TPEX) and terminally exhausted (TEX) cells. Integration of T-cell receptor sequencing data and use of the predicTCR classifier suggest an enrichment of CLL-reactive T cells in lymph nodes. Interactome studies reveal potential immunotherapy targets, notably galectin-9, a TIM3 ligand. Inhibiting galectin-9 in mice reduces disease progression and TIM3+ T cells. Galectin-9 expression also correlates with worse survival in CLL and other cancers, suggesting its role in immune evasion and potential as a therapeutic target.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: D.S. reports funding from GSK and received fees/honoraria from Immunai, Noetik, Alpenglow and Lunaphore. K.B. reports fees from Lunaphore. C.L.T. and E.W.G. hold patents and PCT applications describing methods to identify tumor-reactive T cells, and are founders or employees of Tcelltech GmbH. All other authors declare no competing interests.
Figures

Similar articles
-
Multi-omics profiling identifies TNFRSF18 as a novel marker of exhausted CD8⁺ T cells and reveals tumour-immune dynamics in colorectal cancer.Clin Transl Med. 2025 Aug;15(8):e70425. doi: 10.1002/ctm2.70425. Clin Transl Med. 2025. PMID: 40770837 Free PMC article.
-
Galectin-9 and Tim-3 in gastric cancer: a checkpoint axis driving T cell exhaustion and Treg-mediated immunosuppression independently of anti-PD-1 blockade.Front Immunol. 2025 Jul 1;16:1600792. doi: 10.3389/fimmu.2025.1600792. eCollection 2025. Front Immunol. 2025. PMID: 40666515 Free PMC article.
-
Activated Galectin-9/Tim3 promotes Treg and suppresses Th1 effector function in chronic lymphocytic leukemia.FASEB J. 2021 Jul;35(7):e21556. doi: 10.1096/fj.202100013R. FASEB J. 2021. PMID: 34137463
-
Mixed-methods research to support the use of new lymphoma-specific patient-reported symptom measures derived from the EORTC item library.J Patient Rep Outcomes. 2024 Jan 22;8(1):8. doi: 10.1186/s41687-024-00683-2. J Patient Rep Outcomes. 2024. PMID: 38252198 Free PMC article. Review.
-
Clinical Practice Recommendations for Use of Allogeneic Hematopoietic Cell Transplantation in Chronic Lymphocytic Leukemia on Behalf of the Guidelines Committee of the American Society for Blood and Marrow Transplantation.Biol Blood Marrow Transplant. 2016 Dec;22(12):2117-2125. doi: 10.1016/j.bbmt.2016.09.013. Epub 2016 Sep 19. Biol Blood Marrow Transplant. 2016. PMID: 27660167 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
