

CXCL16 Producing Tumor Clones Are Shaping Immunosuppressive Microenvironment in Squamous Cell Carcinoma via CXCR6 Regulatory T Cell
- PMID: 40776410
- PMCID: PMC12331524
- DOI: 10.1002/cam4.71060
CXCL16 Producing Tumor Clones Are Shaping Immunosuppressive Microenvironment in Squamous Cell Carcinoma via CXCR6 Regulatory T Cell
Abstract
Background: Cutaneous squamous cell carcinoma (cSCC) and basal cell carcinoma (BCC) are the most prevalent types of nonmelanoma skin cancer (NMSC) and exhibit significant inter- and intra-tumor heterogeneity. cSCC has a higher metastatic potential than BCC, accompanied by a considerable mortality rate. However, the detailed mechanisms of tumor evolution in cSCC have not yet been described.
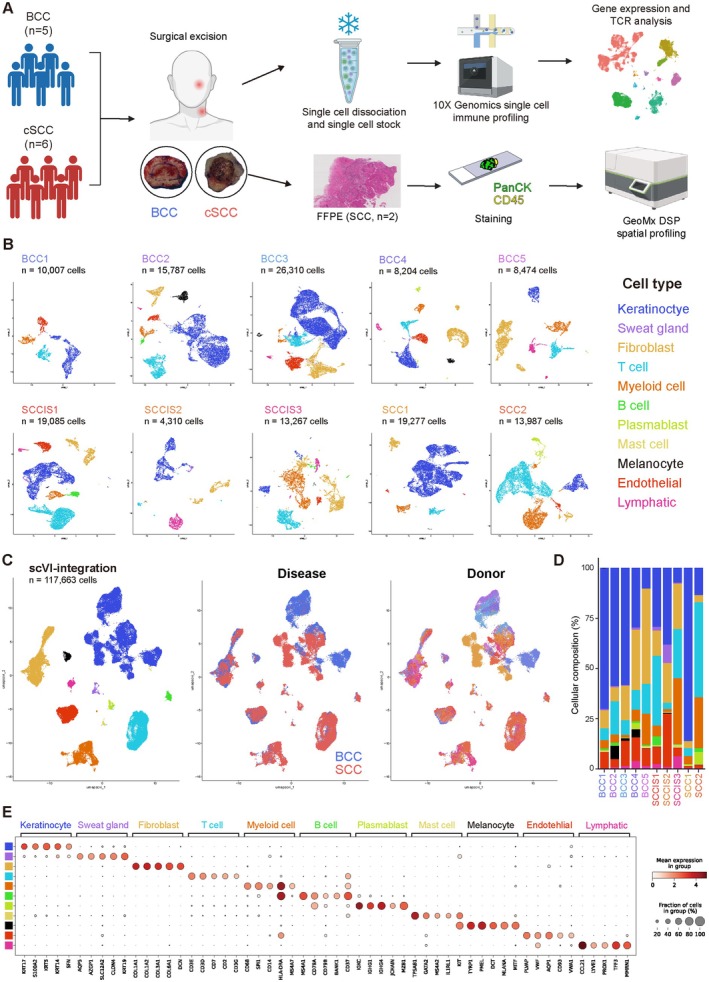
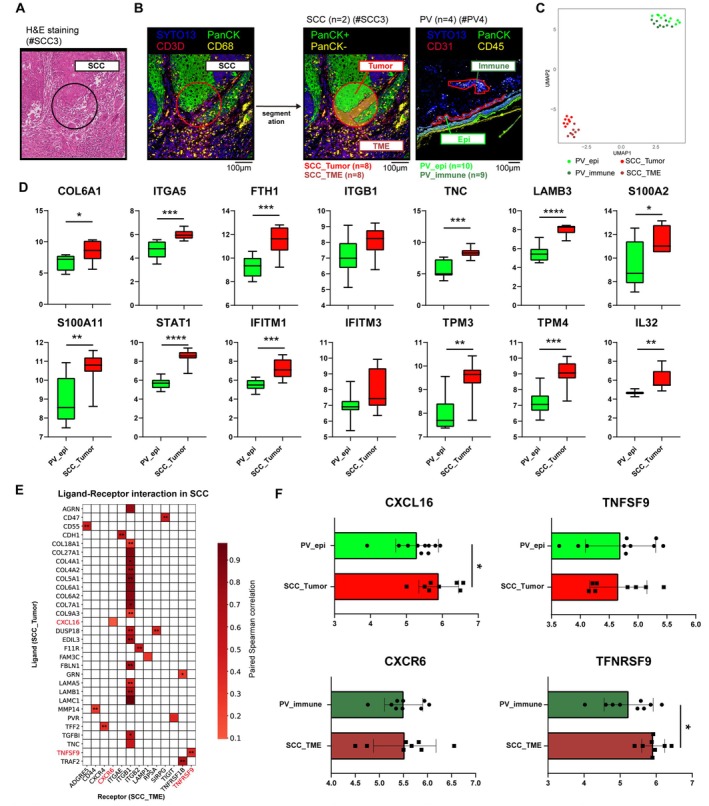
Methods: We performed single-cell RNA sequencing (scRNA-seq) and T cell receptor (TCR) clonal analysis of skin biopsies from five BCCs, three squamous cell carcinomas in situ (SCCIS), and two invasive squamous cell carcinomas (SCC). Independent SCC specimens were used for spatial transcriptomic (ST) analysis using GeoMx Digital Spatial Profiler (DSP).
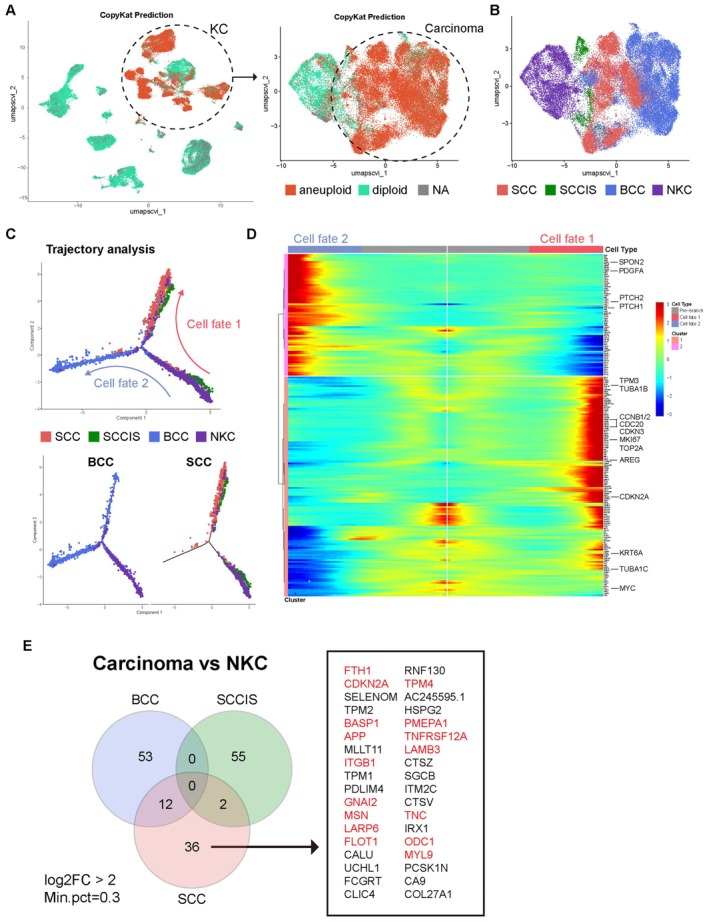
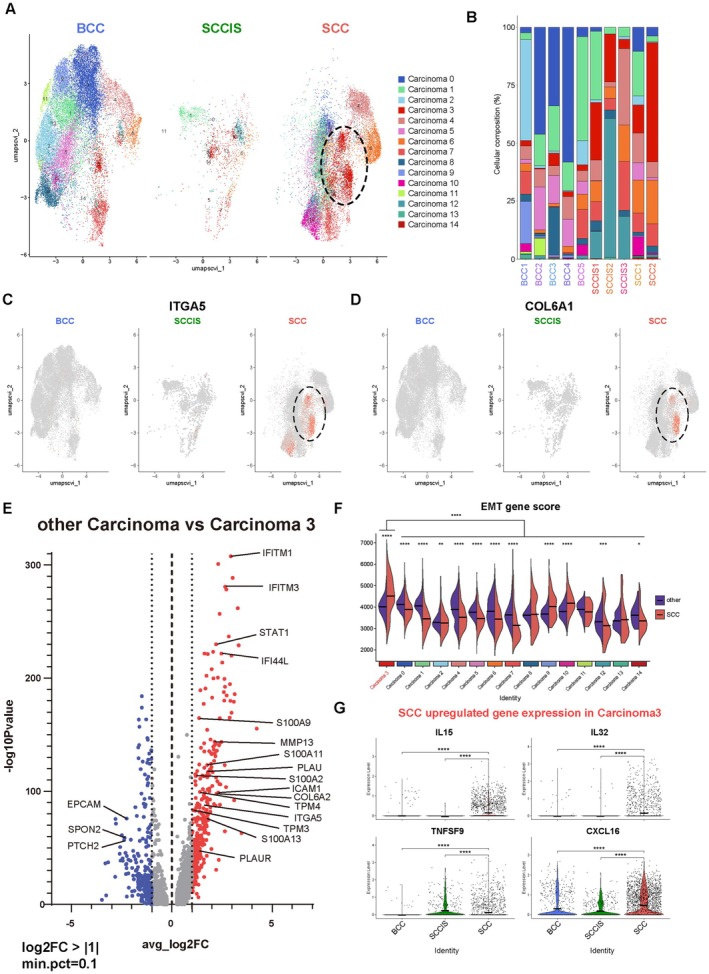
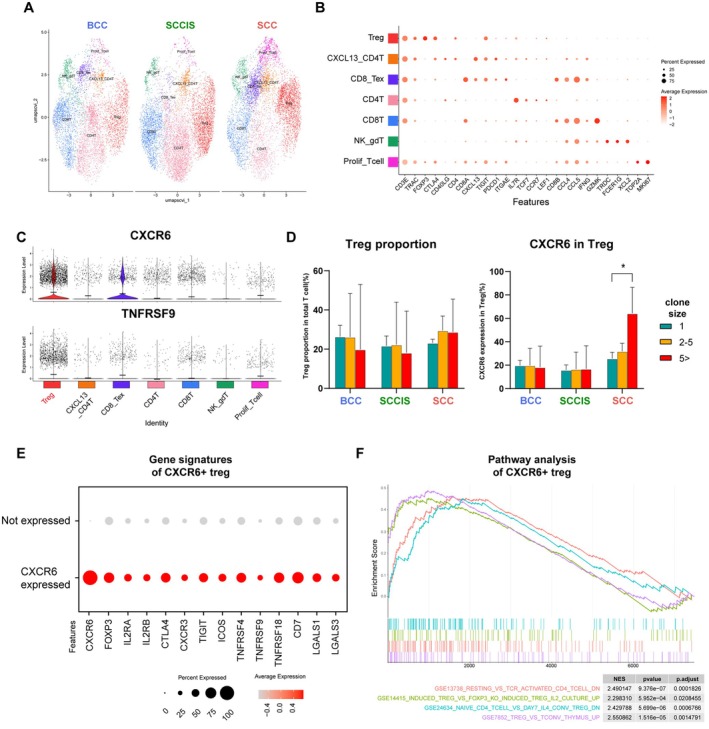
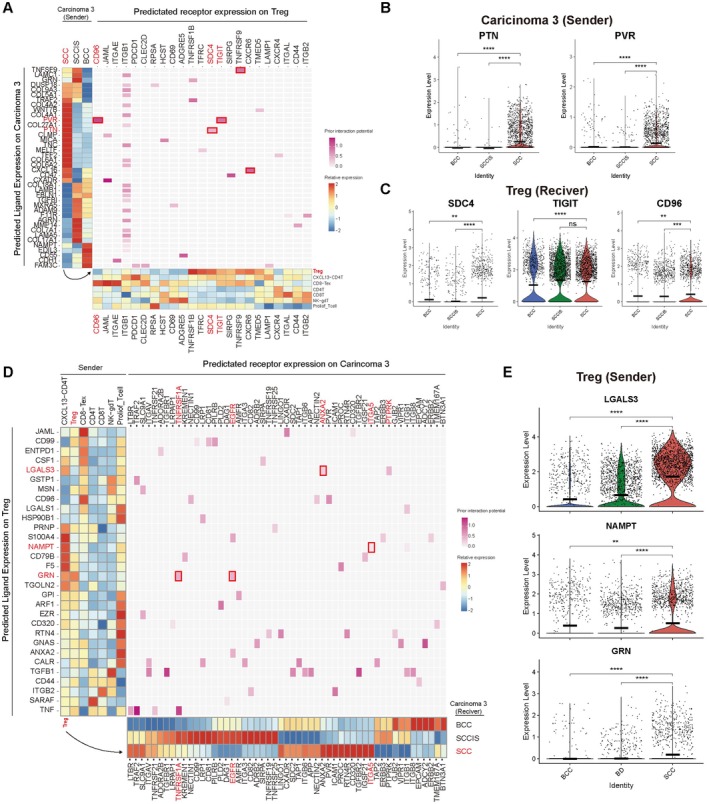
Result: Using scRNA-seq, we analyzed a total of 117,663 cells. We distinguished cancer cells using copy number variation and identified SCC-specific genes that potentially contribute to tumor progression. Analysis of tumor clones revealed SCC-specific COL6A1+/ITGA5+ carcinoma cells which produce CXCL16. We also annotated CXCR6+ regulatory T cells (Tregs) which potentially move toward the tumor site by CXCL16, shaping the immunosuppressive TME. ST analysis supported these clones were located at the invasion site of SCC.
Conclusion: We suggest COL6A1 and ITGA5 promote the invasive and metastatic property of SCC. We also uncovered how SCC recruits Tregs via the CXCL16/CXCR6 axis to create a TME favorable for its survival. These molecules can be used as potential therapeutic targets for treatment of SCC.
Keywords: basal cell carcinoma; cutaneous squamous cell carcinoma; scRNA and TCR seq; spatial transcriptomic.
© 2025 The Author(s). Cancer Medicine published by John Wiley & Sons Ltd.
Conflict of interest statement
Integration analysis of scRNA‐seq and ST of NMSC reveals mechanisms by which specific SCC clusters promote metastasis and shape an immunosuppressive TME, providing new approaches for targeted therapies to prevent SCC progression.
The authors declare no conflicts of interest.
Figures
Similar articles
-
St Andrews Referral Delay in Skin Cancer (StARDISC): a study of keratinocyte skin cancer time to treatment, growth, invasiveness, British Association of Dermatologists risk factors and excision adequacy.Br J Dermatol. 2025 Jun 20;193(1):157-166. doi: 10.1093/bjd/ljaf097. Br J Dermatol. 2025. PMID: 40080699
-
Chondroitin sulfate proteoglycan 4 increases invasion of recessive dystrophic epidermolysis bullosa-associated cutaneous squamous cell carcinoma by modifying transforming growth factor-β signalling.Br J Dermatol. 2024 Dec 23;192(1):104-117. doi: 10.1093/bjd/ljae295. Br J Dermatol. 2024. PMID: 39018437 Free PMC article.
-
Sun protection for preventing basal cell and squamous cell skin cancers.Cochrane Database Syst Rev. 2016 Jul 25;7(7):CD011161. doi: 10.1002/14651858.CD011161.pub2. Cochrane Database Syst Rev. 2016. PMID: 27455163 Free PMC article.
-
Pannexin 1 and pannexin 3 differentially regulate the cancer cell properties of cutaneous squamous cell carcinoma.J Physiol. 2025 Aug;603(15):4409-4432. doi: 10.1113/JP286172. Epub 2024 Nov 19. J Physiol. 2025. PMID: 39560179 Free PMC article.
-
Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: a systematic review.Arch Dermatol. 2009 Dec;145(12):1431-8. doi: 10.1001/archdermatol.2009.291. Arch Dermatol. 2009. PMID: 20026854
References
MeSH terms
Substances
Grants and funding
- 800-20230004/the Collaborative Research Program of SNU Boramae Medical Center and Basic Medical Science from Seoul National University College of Medicine
- 800-20230005/the Collaborative Research Program of SNU Boramae Medical Center and Basic Medical Science from Seoul National University College of Medicine
- RS-2024-00512120/Ministry of Health & Welfare, Republic of Korea
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
