

The Clinical and Genetic Landscape of a French Multicenter Cohort of 2563 Epilepsy Patients Referred for Genetic Diagnosis
- PMID: 40778729
- PMCID: PMC12332890
- DOI: 10.1111/ene.70324
The Clinical and Genetic Landscape of a French Multicenter Cohort of 2563 Epilepsy Patients Referred for Genetic Diagnosis
Abstract
Background: Epileptic disorders are a heterogeneous group of neurological conditions, with many cases linked to monogenic causes, particularly in developmental and epileptic encephalopathies (DEE). Identifying pathogenic variants aids treatment, prognosis, and family planning. In France, genetic testing is coordinated through the EpiGene network.
Methods: We analyzed clinical and genetic data from 2563 epilepsy patients referred to four diagnostic labs (2016-2023). Epilepsy syndromes were classified via pre-test questionnaires, and genotyping used various gene panels, including a 68-gene core panel. Multivariate logistic regression assessed diagnostic rates and genotype-phenotype correlations.
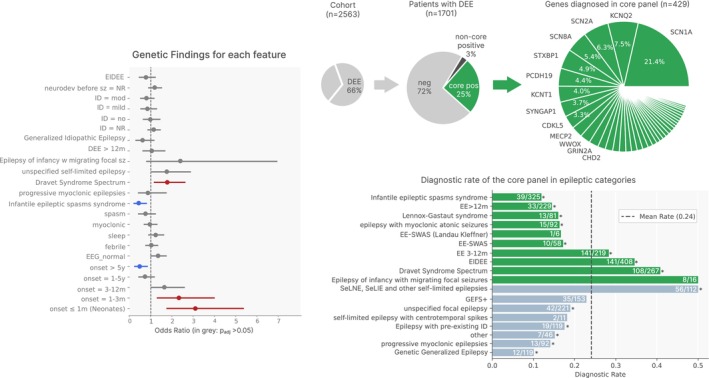
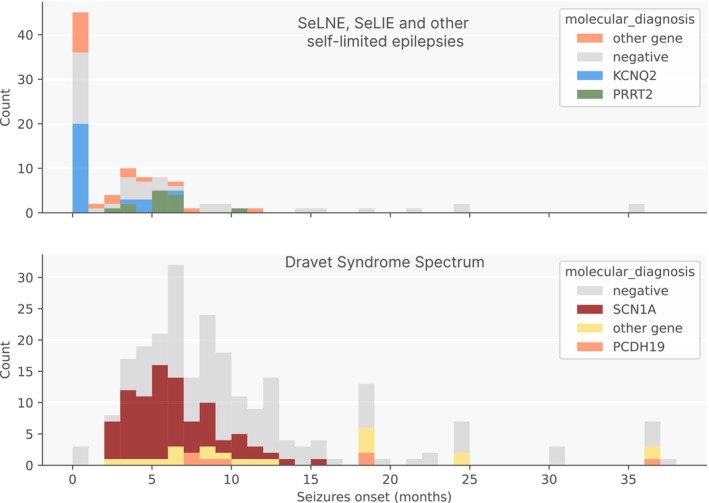
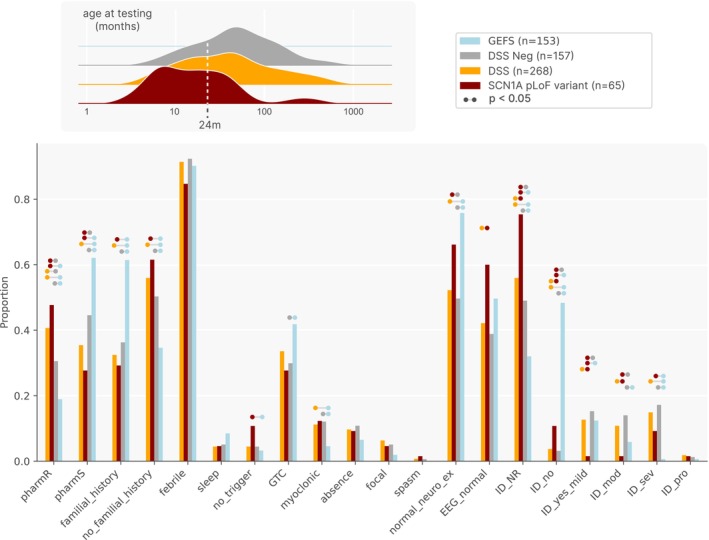
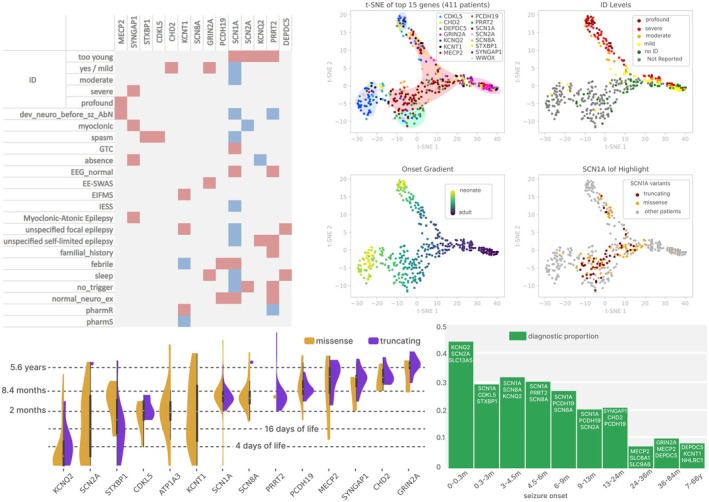
Results: Overall, 27.0% of patients had pathogenic/likely pathogenic variants, mainly within the core panel (24%). SCN1A and KCNQ2 were the most frequently mutated genes. Diagnostic yield varied by syndrome, with Dravet Syndrome Spectrum (DSS) and early-infantile DEE (EIDEE) showing the highest rates (41% and 34%, respectively). Genetic heterogeneity differed across syndromes, from DSS (predominantly SCN1A) to Infantile Epileptic Spasms Syndrome (IESS, 12%), involving ≥ 26 genes. Outside DEE, self-limited neonatal epilepsy (SeLNE) had the highest yield (50%). Earlier seizure onset was associated with a higher likelihood of a positive molecular diagnosis, whereas intellectual disability severity and drug resistance were not independently predictive of diagnostic outcome. Genotype-phenotype correlations highlighted that objective clinical data (e.g., age of onset) can outperform syndrome labels (e.g., EIDEE) in predicting diagnosis.
Conclusion: This large cohort study refines the genetic landscape of epilepsy, informs classification challenges, and enhances genetic testing strategies, ultimately improving patient care and future research directions.
Keywords: Dravet syndrome; Lennox Gastaut syndrome; Spasms, infantile; genetic testing; genetics, medical.
© 2025 The Author(s). European Journal of Neurology published by John Wiley & Sons Ltd on behalf of European Academy of Neurology.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Scheffer I. E., Zuberi S., Mefford H. C., Guerrini R., and McTague A., “Developmental and Epileptic Encephalopathies,” Nature Reviews Disease Primers 10, no. 1 (2024): 61. - PubMed
-
- Millevert C., Weckhuysen S., and For the ILAE Genetics Commission , “ILAE Genetic Literacy Series: Self‐Limited Familial Epilepsy Syndromes With Onset in Neonatal Age and Infancy,” Epileptic Disorders 25, no. 4 (2023): 445–453. - PubMed
-
- Specchio N., Wirrell E. C., Scheffer I. E., et al., “International League Against Epilepsy Classification and Definition of Epilepsy Syndromes With Onset in Childhood: Position Paper by the ILAE Task Force on Nosology and Definitions,” Epilepsia 63, no. 6 (2022): 1398–1442. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
