

Chromatin-dependent motif syntax defines differentiation trajectories
- PMID: 40780181
- PMCID: PMC12488066
- DOI: 10.1016/j.molcel.2025.07.005
Chromatin-dependent motif syntax defines differentiation trajectories
Abstract
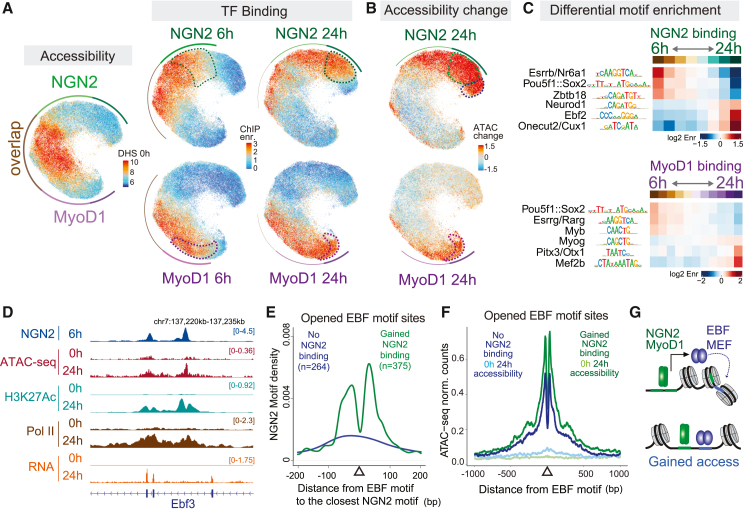
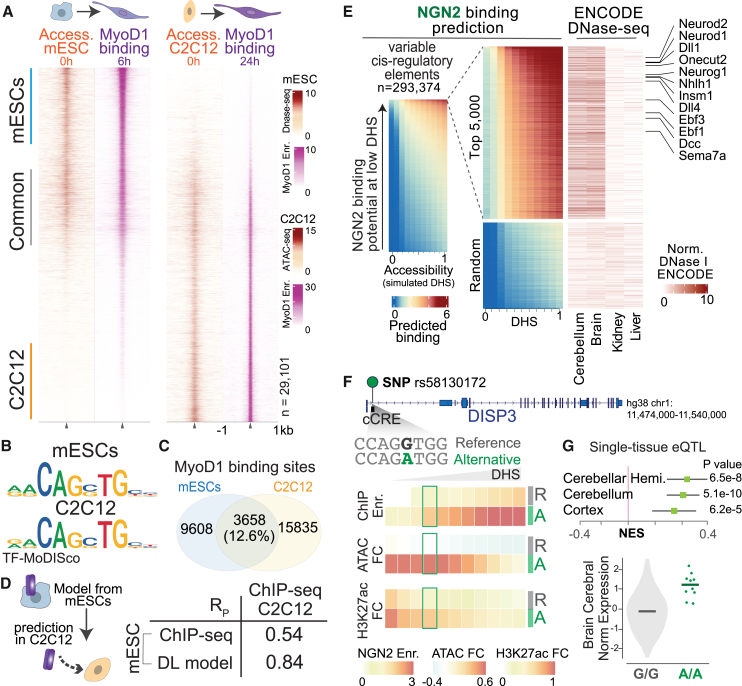
Transcription factors (TFs) recognizing DNA motifs within regulatory regions drive cell identity. Despite recent advances, their specificity remains incompletely understood. Here, we address this by contrasting two TFs, Neurogenin-2 (NGN2) and MyoD1, which recognize ubiquitous E-box motifs yet drive distinct cell fates toward neurons and muscles, respectively. Upon induction in mouse embryonic stem cells, we monitor binding across differentiation, employing an interpretable machine learning approach that integrates preexisting DNA accessibility. This reveals a chromatin-dependent motif syntax, delineating both common and factor-specific binding, validated by cellular and in vitro assays. Shared binding sites reside in open chromatin, locally influenced by nucleosomes. In contrast, factor-specific binding in closed chromatin involves NGN2 and MyoD1 acting as pioneer factors, influenced by motif variant frequencies, motif spacing, and interaction partners, which together account for subsequent lineage divergence. Transferring our methodology to other models demonstrates how a combination of opportunistic binding and context-specific chromatin-opening underpin TF specificity, driving differentiation trajectories.
Keywords: E-box; cell differentiation; chromatin accessibility; gene regulation; machine learning; motif syntax; motif variants; pioneer factors; predictive models; transcription factor specificity.
Copyright © 2025 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures






References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
