

NOX-NOS crosstalk in the liver-brain axis: Novel insights for redox regulation and neurodegenerative diseases
- PMID: 40782521
- PMCID: PMC12356045
- DOI: 10.1016/j.redox.2025.103807
NOX-NOS crosstalk in the liver-brain axis: Novel insights for redox regulation and neurodegenerative diseases
Abstract
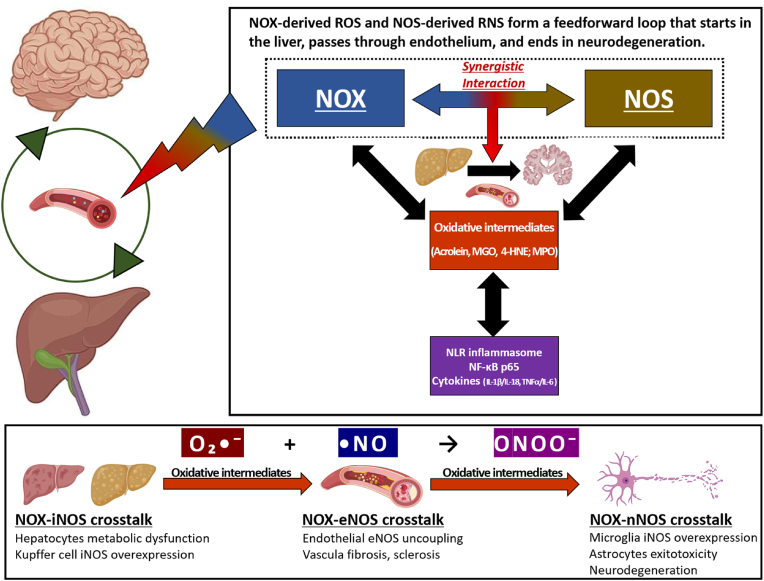
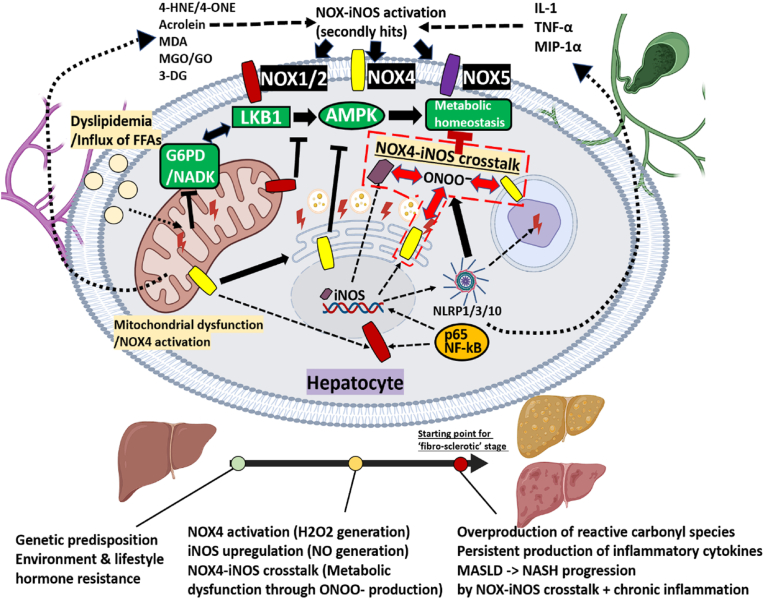
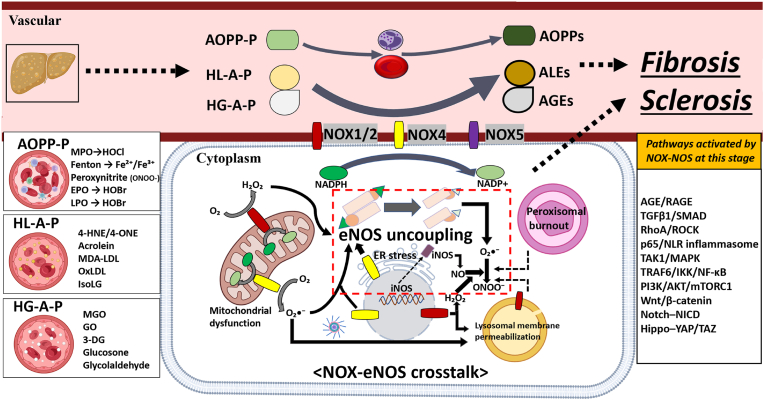
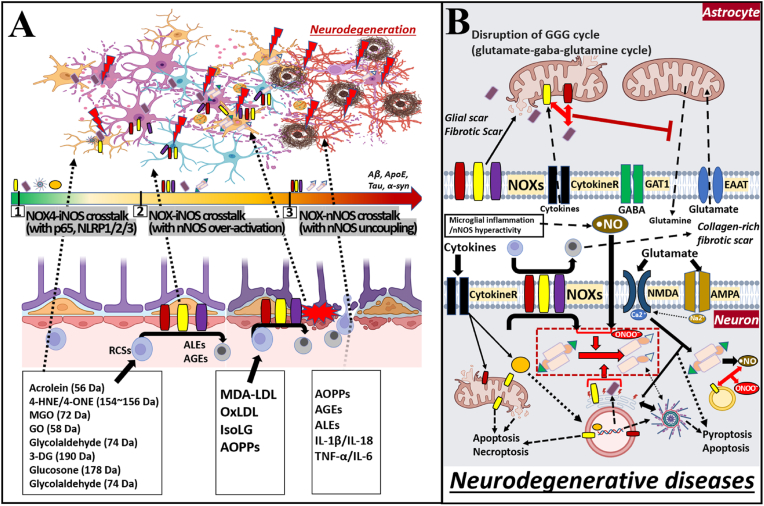
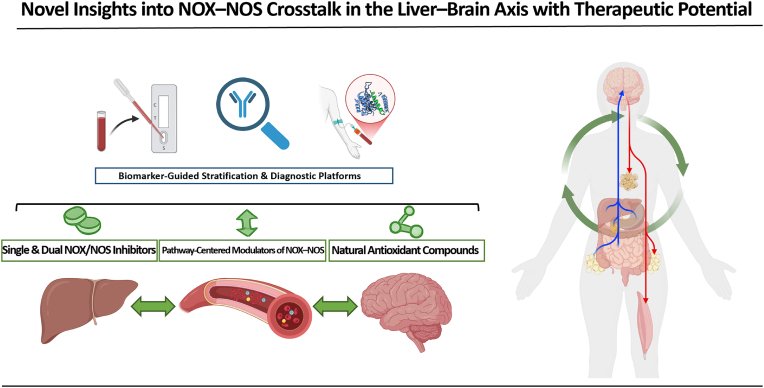
The liver-brain axis is an emerging concept linking liver dysfunction and brain disease. Hepatic metabolic abnormalities induce systemic oxidative stress and endothelial dysfunction, which contribute to central nervous system (CNS) inflammation and neurodegeneration. Redox regulation plays a key role in the liver-brain axis, with NADPH oxidase (NOX) and nitric oxide synthase (NOS) being involved in the generation of various reactive oxygen species (ROS) and reactive nitrogen species (RNS), respectively, thereby inducing oxidative stress and disrupting the NADPH/NADP balance. Dysregulation of NOX-NOS cross-signaling not only amplifies oxidative stress, but also disrupts endothelial homeostasis and exacerbates neuroinflammation, leading to progressive neurodegeneration. For instance, reactive carbonyl species such as methylglyoxal (MGO) and acrolein can upregulate NOX isoforms and stimulate NLRP (NOD like receptor protein) inflammasomes activation, illustrating disease-relevant links between hepatic redox imbalance and CNS pathology. Mechanistically, superoxide (O2•-) generated by NOX readily reacts with nitric oxide (•NO) derived from NOS to form peroxynitrite (ONOO-), a highly reactive oxidant that exacerbates vascular and neuronal injury. Despite extensive research on NOX and NOS, their interactive contributions to redox imbalance and the progression of neurodegenerative diseases remain poorly understood. In this review, we introduce the NOX-NOS axis as a key regulator of the liver-brain axis, and highlight the roles of NOX and NOS in linking hepatic metabolic dysfunction to central nervous system pathology through intermediary metabolites in the exacerbation of neuroinflammation and oxidative stress. We also explore therapeutic strategies targeting NOX-NOS interactions, including selective NOX inhibitors, NOS modulators, and redox homeostasis regulators, providing new insights into redox regulation and the management of metabolic neurodegenerative diseases.
Keywords: Liver-brain axis; NADPH oxidase; NOX-NOS crosstalk; Neurodegenerative diseases; Nitric oxide synthase; Oxidation-reduction reaction.
Copyright © 2025 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures

References
-
- Riaz Z., Richardson G.S., Jin H., Zenitsky G., Anantharam V., Kanthasamy A., Kanthasamy A.G. Nuclear pore and nucleocytoplasmic transport impairment in oxidative stress-induced neurodegeneration: relevance to molecular mechanisms in Pathogenesis of Parkinson's and other related neurodegenerative diseases. Mol. Neurodegener. 2024;19(1):87. doi: 10.1186/s13024-024-00774-0. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
