

Quality Evaluation Based Simulation Selection (QEBSS) for analysis of conformational ensembles and dynamics of multidomain proteins
- PMID: 40783454
- PMCID: PMC12335556
- DOI: 10.1038/s42004-025-01623-x
Quality Evaluation Based Simulation Selection (QEBSS) for analysis of conformational ensembles and dynamics of multidomain proteins
Abstract
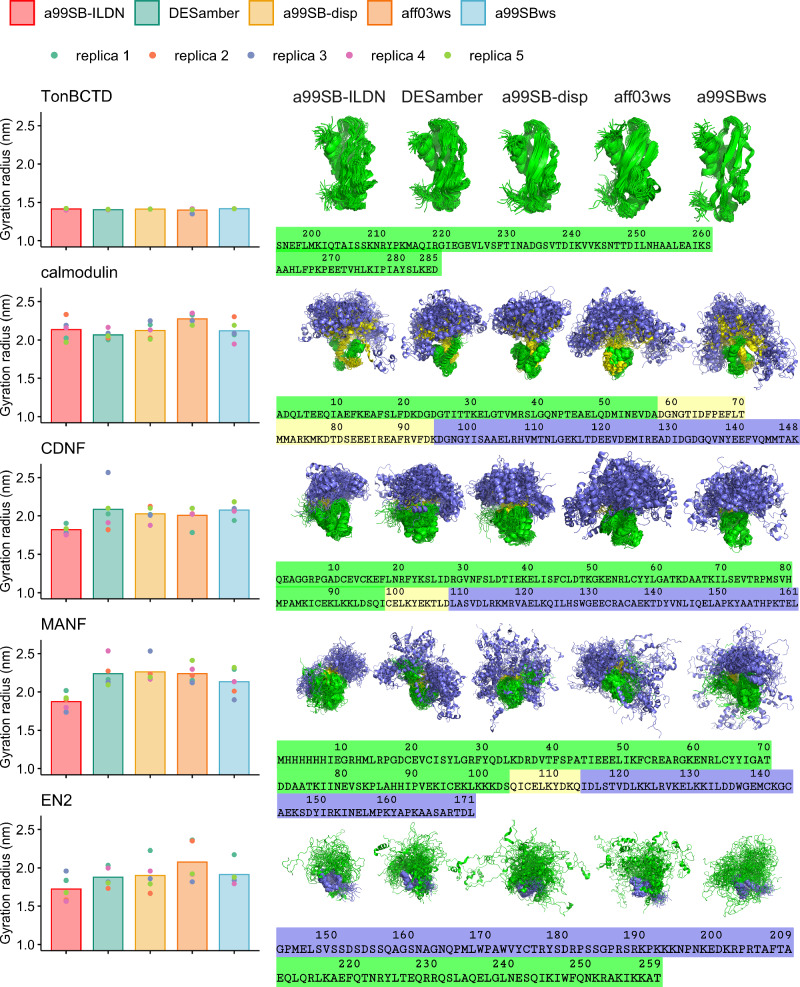
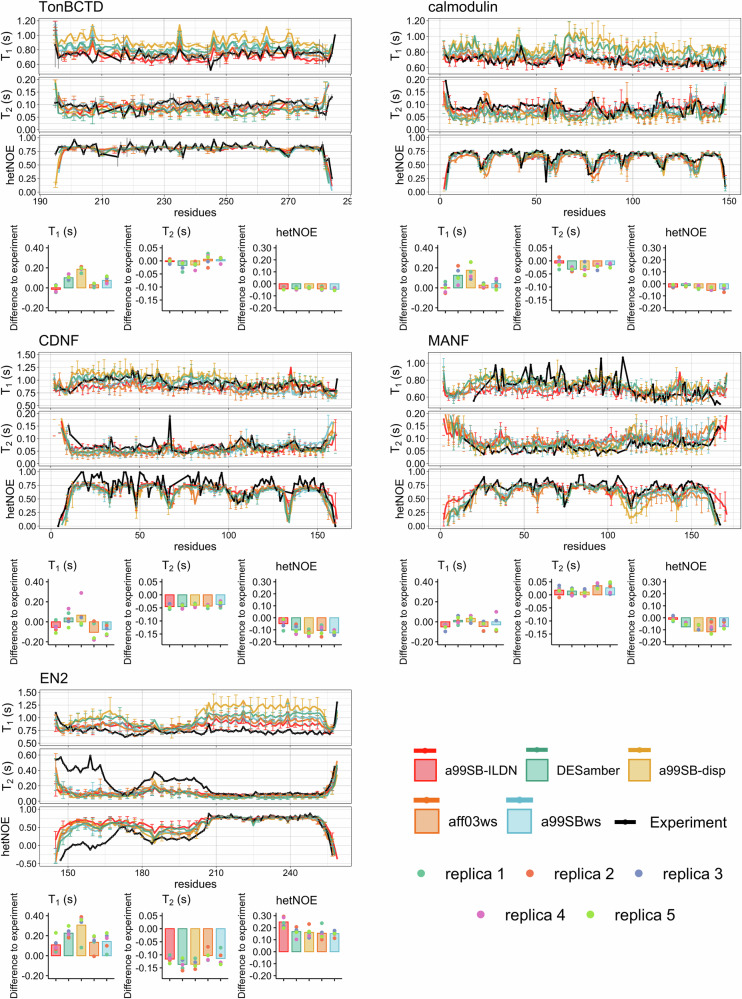
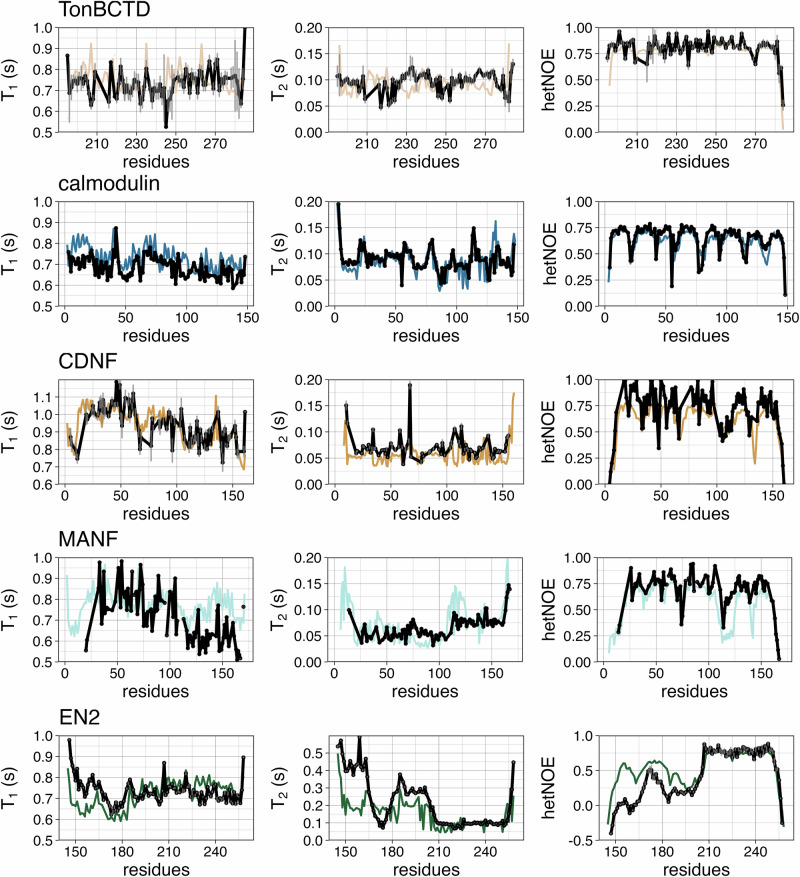
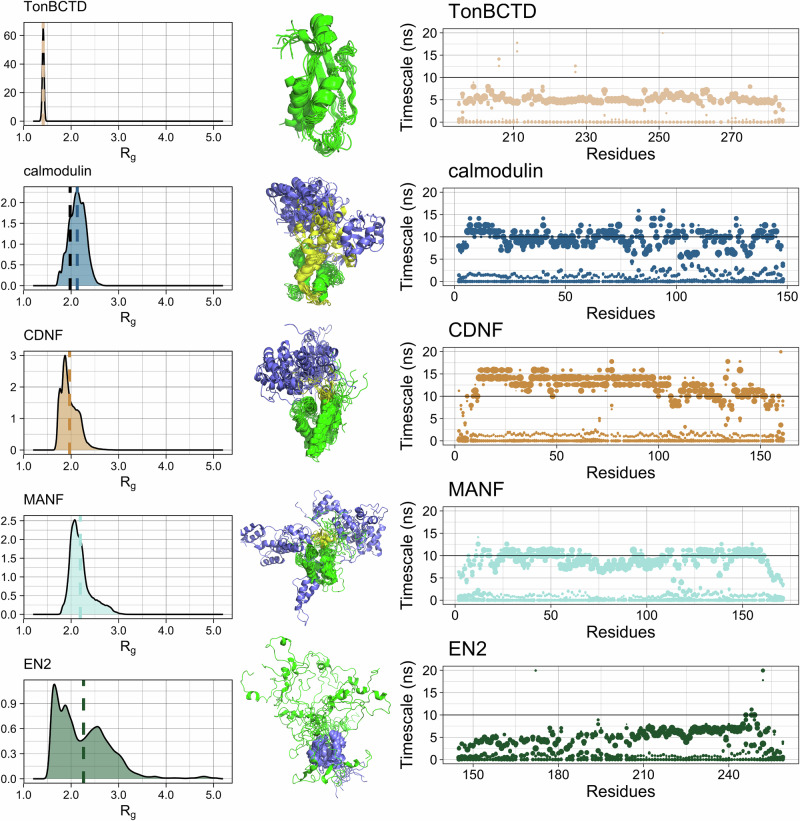
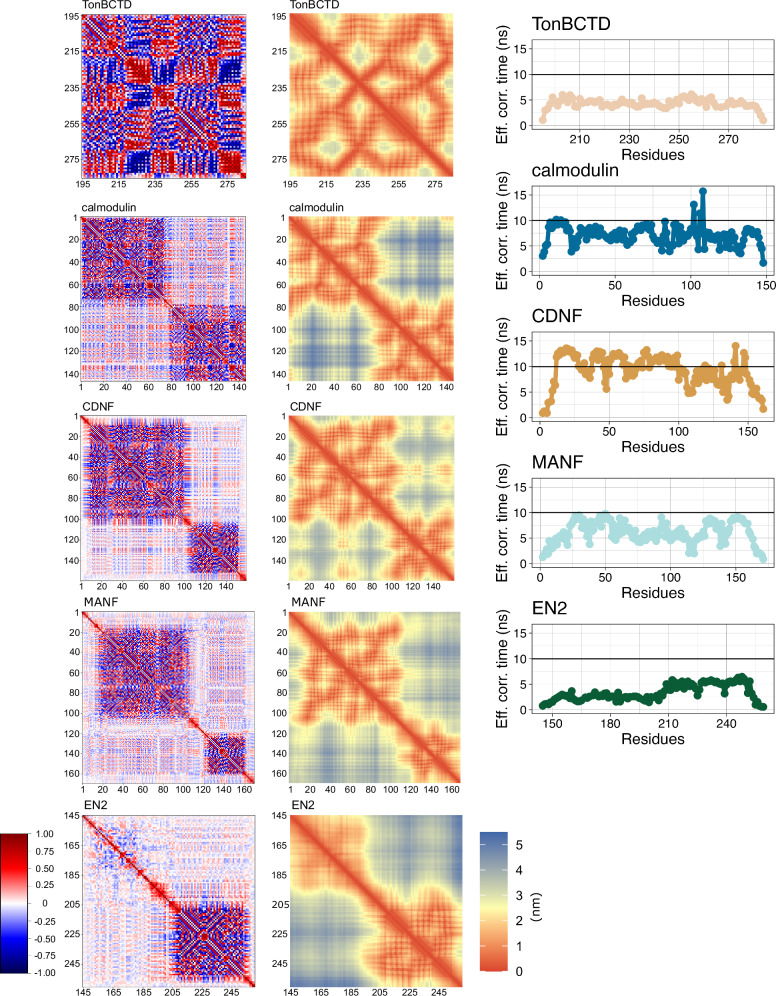
Multidomain proteins containing both folded and intrinsically disordered regions are crucial for biological processes, but characterizing their conformational ensembles and dynamics remains challenging. We introduce the Quality Evaluation Based Simulation Selection (QEBSS) protocol, which combines MD simulations with NMR-derived protein backbone 15N T1 and T2 spin relaxation times and hetNOE values to interpret conformational ensembles and dynamics of multidomain proteins. We demonstrate the practical advantage of QEBSS by characterizing four flexible multidomain proteins: calmodulin, EN2, MANF, and CDNF. These biologically important proteins have been difficult to study due to their flexible nature. Our findings reveal new insights into their conformational landscapes and dynamics, providing mechanistic understanding of their biological functions. QEBSS offers quantitative quality evaluation of simulations and a systematic approach for resolving conformational ensembles of multidomain proteins with heterogeneous dynamics. Given the importance of such proteins in biology, biotechnology, and materials science, QEBSS should benefit fields from drug design to novel materials development.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
Management of urinary stones by experts in stone disease (ESD 2025).Arch Ital Urol Androl. 2025 Jun 30;97(2):14085. doi: 10.4081/aiua.2025.14085. Epub 2025 Jun 30. Arch Ital Urol Androl. 2025. PMID: 40583613 Review.
-
Smartphone and tablet self management apps for asthma.Cochrane Database Syst Rev. 2013 Nov 27;2013(11):CD010013. doi: 10.1002/14651858.CD010013.pub2. Cochrane Database Syst Rev. 2013. PMID: 24282112 Free PMC article.
-
Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children.Cochrane Database Syst Rev. 2012 Jan 18;1:CD008965. doi: 10.1002/14651858.CD008965.pub3. Cochrane Database Syst Rev. 2012. Update in: Cochrane Database Syst Rev. 2014 Apr 10;(4):CD008965. doi: 10.1002/14651858.CD008965.pub4. PMID: 22258996 Updated.
-
Structural dynamics of IDR interactions in human SFPQ and implications for liquid-liquid phase separation.Acta Crystallogr D Struct Biol. 2025 Jul 1;81(Pt 7):357-379. doi: 10.1107/S2059798325005303. Epub 2025 Jun 27. Acta Crystallogr D Struct Biol. 2025. PMID: 40574713 Free PMC article.
Cited by
-
Structure-Based Experimental Datasets for Benchmarking Protein Simulation Force Fields [Article v0.1].ArXiv [Preprint]. 2025 Mar 25:arXiv:2303.11056v2. ArXiv. 2025. PMID: 40196146 Free PMC article. Preprint.
References
-
- Vogel, C., Bashton, M., Kerrison, N. D., Chothia, C. & Teichmann, S. A. Structure, function and evolution of multidomain proteins. Curr. Opin. Struct. Biol.14, 208–16 (2004). - PubMed
-
- Shapiro, Y. E. Nmr spectroscopy on domain dynamics in biomacromolecules. Prog. Biophys. Mol. Biol.112, 58–117 (2013). - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
