

The role of environmentally mediated drug resistance in facilitating the spatial distribution of residual disease
- PMID: 40783466
- PMCID: PMC12335614
- DOI: 10.1038/s42003-025-08585-9
The role of environmentally mediated drug resistance in facilitating the spatial distribution of residual disease
Abstract
The development of de novo resistance is a major disadvantage in molecularly targeted therapies. While much focus is on cell-intrinsic mechanisms, the microenvironment is also known to play a crucial role. This study examines interactions between cancer cells and cancer associated fibroblasts (CAFs) to understand the local crosstalk facilitating residual disease. Using a hybrid-discrete-continuum model, we explore how treatment-induced stress responses can elicit CAF activation and how breaks in treatment allow microenvironment normalisation. We investigate how fluctuating environmental conditions shape the local crosstalk and ultimately drive residual disease. Our experimentally calibrated model identifies environmental and treatment conditions that allow tumour eradication and those that enable survival. We find two distinct mechanisms that underpin residual disease: vasculature-limited drug delivery and CAF-mediated rescue. This work provides a better understanding of the mechanisms that drive the creation of localised residual disease, crucial to informing the development of more effective treatment protocols.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
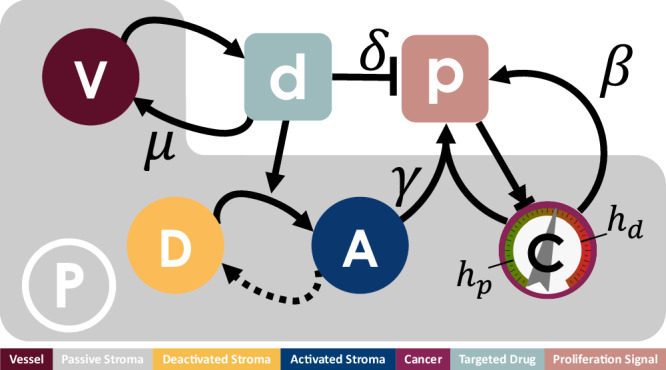
 , behaviour is dependent on the local concentration of the proliferation signal,
, behaviour is dependent on the local concentration of the proliferation signal,  , with thresholds for death, hd, and proliferation, hp. Cancer cells provide autocrine promotion of local proliferation signal at rate β. TME comprises of both passive,
, with thresholds for death, hd, and proliferation, hp. Cancer cells provide autocrine promotion of local proliferation signal at rate β. TME comprises of both passive,  , and reactive stroma. Reactive stroma can be in either an activated,
, and reactive stroma. Reactive stroma can be in either an activated,  , or deactivated,
, or deactivated,  , state. A targeted inhibitor drug,
, state. A targeted inhibitor drug,  , depletes proliferation signal at rate δ and is removed from the system through vessel sites
, depletes proliferation signal at rate δ and is removed from the system through vessel sites  at rate μ. Local concentration of targeted drug above threshold hr triggers activation of reactive stroma cells adjacent to a cancer cell, in turn providing paracrine promotion of the proliferation signal at rate γ. Activated reactive stroma reverts to a deactivated state if the drug concentration falls below hr.
at rate μ. Local concentration of targeted drug above threshold hr triggers activation of reactive stroma cells adjacent to a cancer cell, in turn providing paracrine promotion of the proliferation signal at rate γ. Activated reactive stroma reverts to a deactivated state if the drug concentration falls below hr.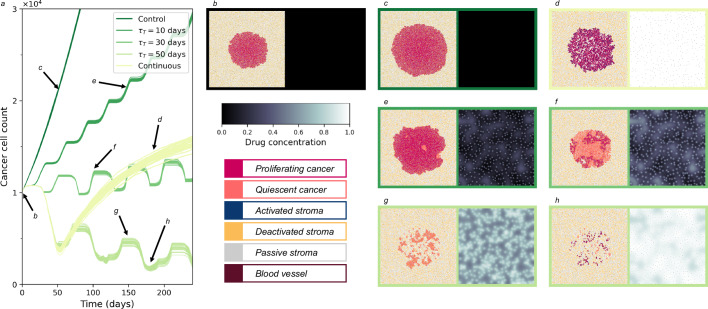
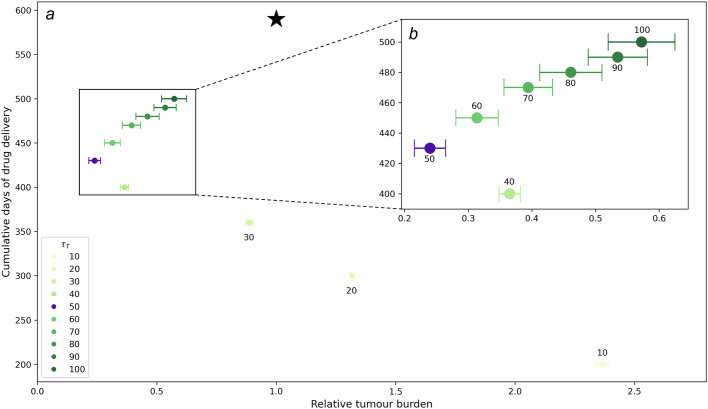
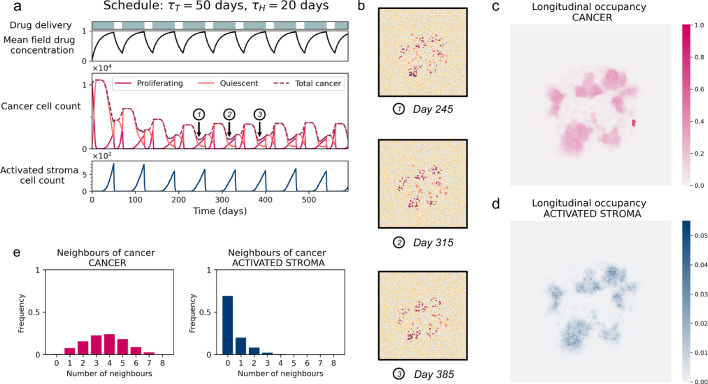
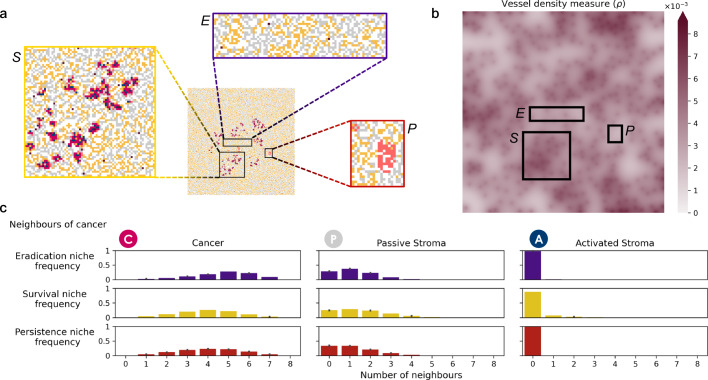
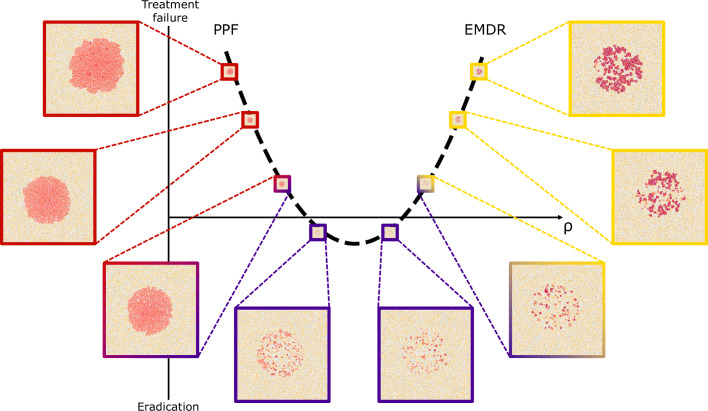
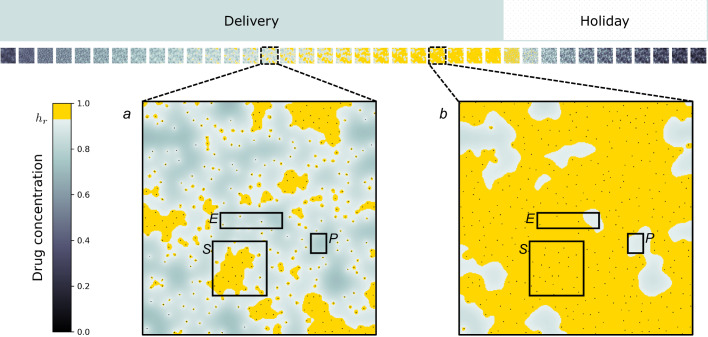
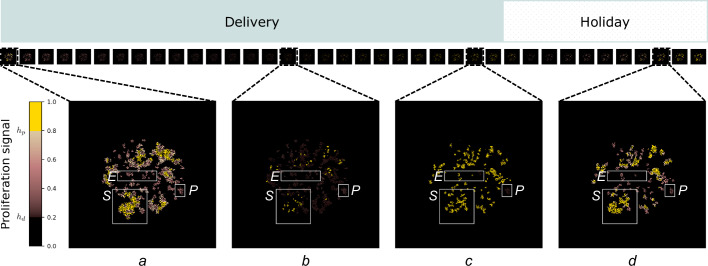
Similar articles
-
The multifaceted contributions of cancer-associated fibroblasts to drug resistance in primary and metastatic tumors.Drug Resist Updat. 2025 Sep;82:101273. doi: 10.1016/j.drup.2025.101273. Epub 2025 Jul 1. Drug Resist Updat. 2025. PMID: 40617187 Review.
-
Hypoxia-induced upregulation of HIF1A-AS3 promotes MSC transition to cancer-associated fibroblasts and confers drug resistance in gastric cancer.Drug Resist Updat. 2025 Sep;82:101275. doi: 10.1016/j.drup.2025.101275. Epub 2025 Jul 14. Drug Resist Updat. 2025. PMID: 40706422
-
Hidden forces: the impact of cancer-associated fibroblasts on non-small cell lung cancer development and therapy.J Transl Med. 2025 Jul 25;23(1):830. doi: 10.1186/s12967-025-06791-x. J Transl Med. 2025. PMID: 40713753 Free PMC article. Review.
-
Drug resistant pancreatic cancer cells exhibit altered biophysical interactions with stromal fibroblasts in imaging studies of 3D co-culture models.Sci Rep. 2024 Sep 5;14(1):20698. doi: 10.1038/s41598-024-71372-9. Sci Rep. 2024. PMID: 39237667 Free PMC article.
-
Impact of residual disease as a prognostic factor for survival in women with advanced epithelial ovarian cancer after primary surgery.Cochrane Database Syst Rev. 2022 Sep 26;9(9):CD015048. doi: 10.1002/14651858.CD015048.pub2. Cochrane Database Syst Rev. 2022. PMID: 36161421 Free PMC article.
References
-
- Bedard, P. L., Hyman, D. M., Davids, M. S. & Siu, L. L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet395, 1078–1088 (2020). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
