

Cathepsin S regulates ferroptosis sensitivity in hepatocellular carcinoma through the KEAP1-NRF2 signaling pathway
- PMID: 40784043
- PMCID: PMC12357265
- DOI: 10.1016/j.redox.2025.103815
Cathepsin S regulates ferroptosis sensitivity in hepatocellular carcinoma through the KEAP1-NRF2 signaling pathway
Abstract
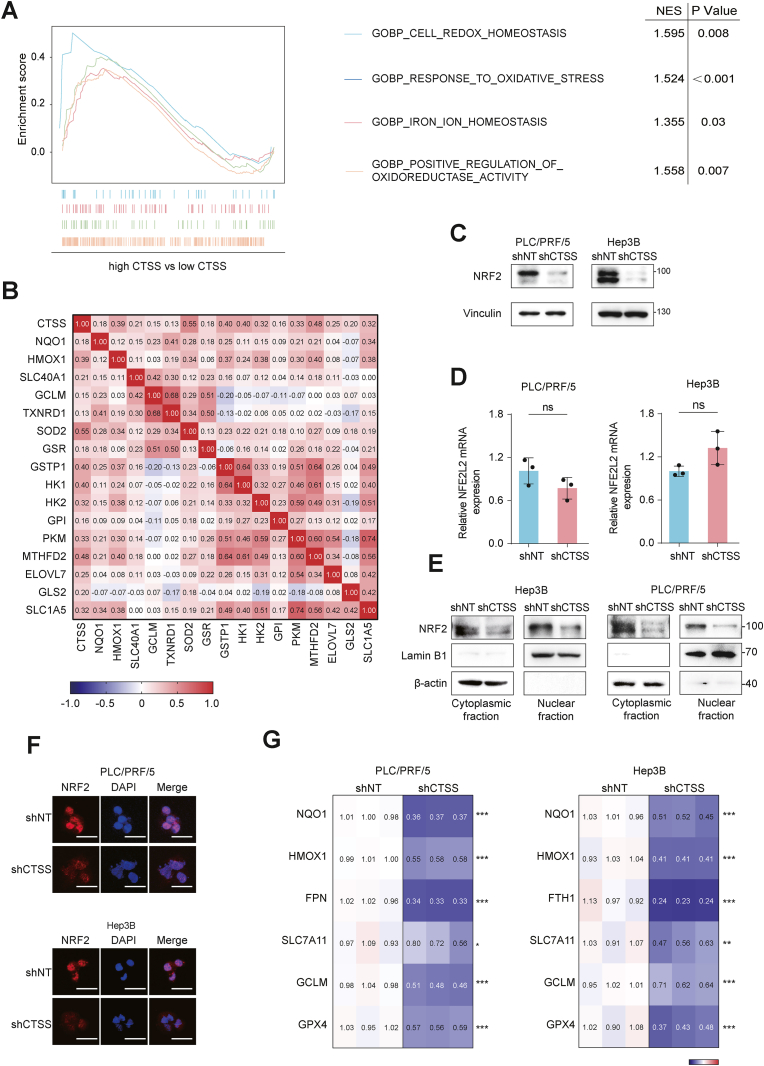
Ferroptosis is a newly discovered iron-dependent programmed cell death characterized by excess lipid peroxidation. It is emerging as a promising target for tumor therapies. In the present study, we first identify Cathepsin S (CTSS) as a novel ferroptosis regulator. CTSS is upregulated in ferroptosis-resistant hepatocellular carcinoma (HCC) cells, and suppression of CTSS sensitizes HCC cells to ferroptosis. Mechanistically, ferroptosis stress induces CTSS maturation and promotes the autophagy-lysosomal degradation of Kelch-like ECH-associated protein 1 (KEAP1). This process blocks KEAP1-dependent, ubiquitination-mediated degradation of nuclear factor E2-related factor 2 (NRF). Consequently, the accumulated NRF2 translocates from the cytoplasm to the nucleus and drives the transcription of anti-ferroptosis genes. In vivo study reveals that CTSS depletion, achieved through either shRNA or the specific inhibitor LY3000328, in combination with a ferroptosis inducer, inhibits HCC tumor growth in orthotopic xenograft mouse models. In conclusion, the above data suggest that CTSS can potentiate ferroptosis in HCC cells and may be a therapeutic target to overcome ferroptosis resistance in HCC patients.
Keywords: CTSS; HCC; Kelch-like ECH-associated protein 1; LY3000328; Nuclear factor E2-related factor 2.
Copyright © 2025 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures








References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
