

Technological advances in the diagnosis and management of inherited optic neuropathies
- PMID: 40786632
- PMCID: PMC12331713
- DOI: 10.3389/fneur.2025.1609033
Technological advances in the diagnosis and management of inherited optic neuropathies
Abstract
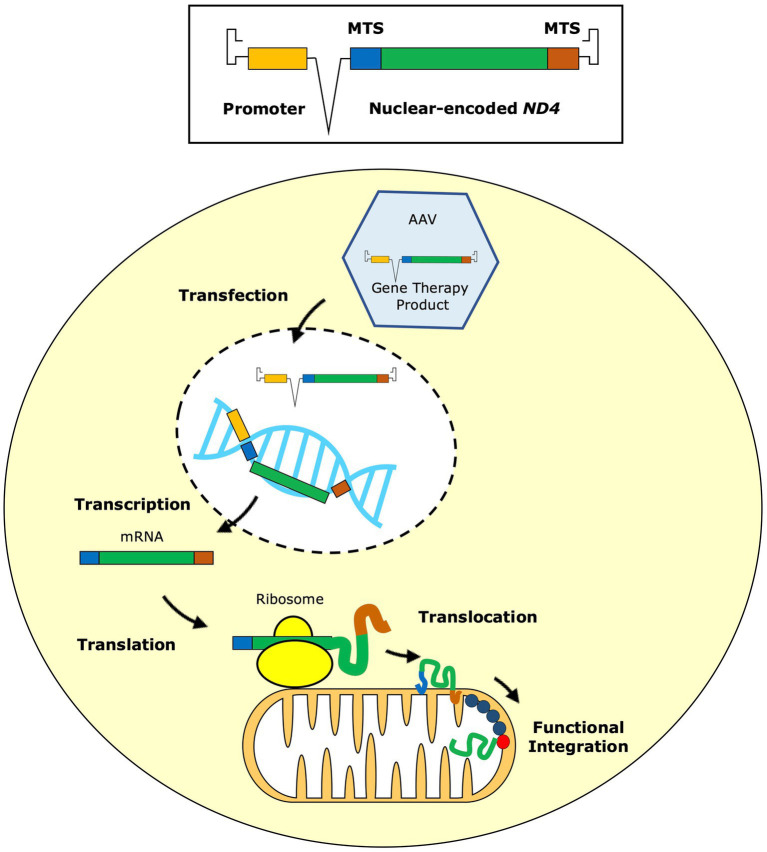
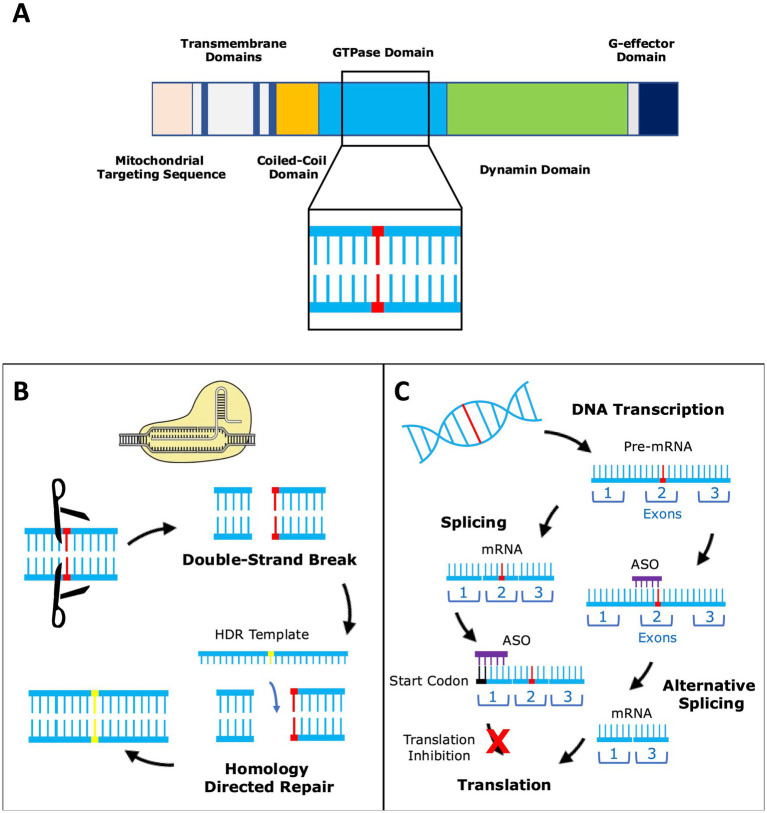
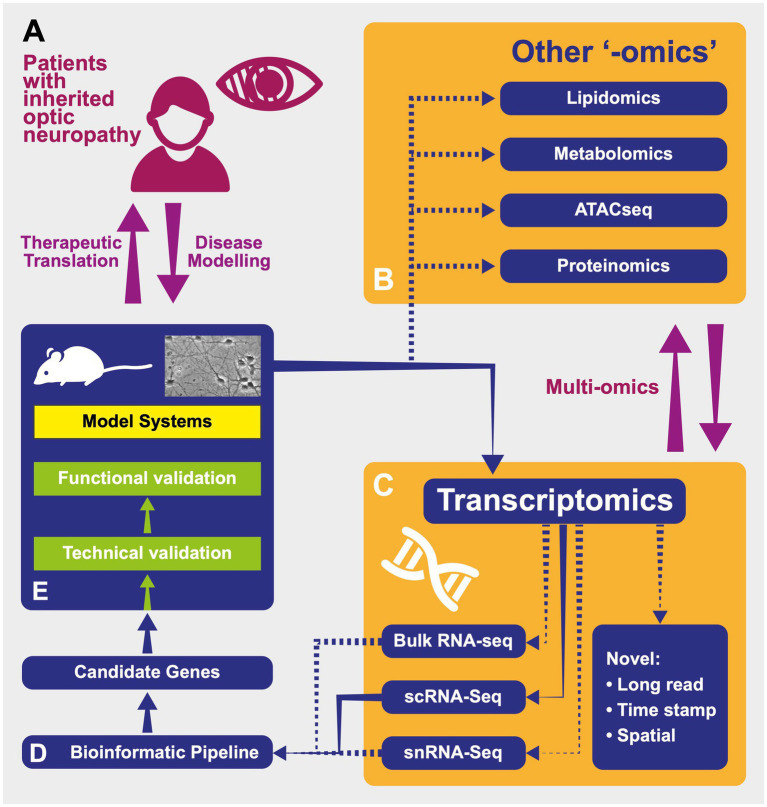
Preferential degeneration of retinal ganglion cells (RGCs) is a defining feature of the inherited optic neuropathies (IONs), a group of monogenic eye diseases predominately comprising Leber hereditary optic neuropathy (LHON) and autosomal dominant optic atrophy (DOA). Their pathogenesis is characterised by mitochondrial dysfunction, which causes loss of RGCs leading to irreversible vision loss. Although currently incurable, there are several emerging therapeutic avenues encompassing gene therapies, precision medicine strategies and neuroprotection. These are underscored by recent technological advances such as next-generation sequencing and improved disease modelling. In this review, we discuss these advances and the impact these will have on future diagnostic and treatment capabilities. We first focus on the clinical presentation and pathogenic mechanisms of LHON and DOA, followed by a discussion of emerging technology to facilitate diagnosis and treatment. We highlight the current unmet clinical demand of IONs, and the promise of current and future research developments.
Keywords: autosomal dominant; hereditary; leber; optic atrophy; retinal ganglion cells.
Copyright © 2025 Britton, Yu-Wai-Man and Chen.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Insights on the Genetic and Phenotypic Complexities of Optic Neuropathies.Genes (Basel). 2024 Nov 29;15(12):1559. doi: 10.3390/genes15121559. Genes (Basel). 2024. PMID: 39766826 Free PMC article.
-
Extraocular features of Leber hereditary optic neuropathy: A scoping review.J Biol Methods. 2025 May 14;12(2):e99010055. doi: 10.14440/jbm.2024.0113. eCollection 2025. J Biol Methods. 2025. PMID: 40787643 Free PMC article. Review.
-
Disruption of mitochondrial homeostasis and permeability transition pore opening in OPA1 iPSC-derived retinal ganglion cells.Acta Neuropathol Commun. 2025 Feb 13;13(1):28. doi: 10.1186/s40478-025-01942-z. Acta Neuropathol Commun. 2025. PMID: 39948685 Free PMC article.
-
Cell-based therapies for traumatic optic neuropathy: Recent advances, challenges, and perspectives.Neural Regen Res. 2025 Jun 19. doi: 10.4103/NRR.NRR-D-24-01322. Online ahead of print. Neural Regen Res. 2025. PMID: 40537004
-
Uncommon Non-MS Demyelinating Disorders of the Central Nervous System.Curr Neurol Neurosci Rep. 2025 Jul 1;25(1):45. doi: 10.1007/s11910-025-01432-8. Curr Neurol Neurosci Rep. 2025. PMID: 40591029 Review.
References
-
- Nissen C, Rönnbäck C, Sander B, Herbst K, Milea D, Larsen M, et al. Dissociation of pupillary post-illumination responses from visual function in confirmed OPA1 c.983A > G and c.2708_2711delTTAG autosomal dominant optic atrophy. Front Neurol. (2015) 6:5. doi: 10.3389/fneur.2015.00005 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources