

This is a preprint.
Neuroligin-3 interaction with CSPG4 regulates normal and malignant glial precursors through PIEZO1
- PMID: 40791371
- PMCID: PMC12338510
- DOI: 10.1101/2025.07.12.664340
Neuroligin-3 interaction with CSPG4 regulates normal and malignant glial precursors through PIEZO1
Abstract
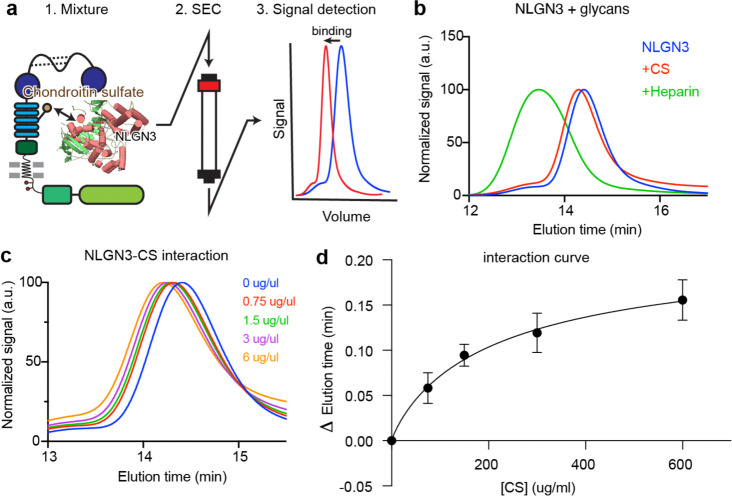
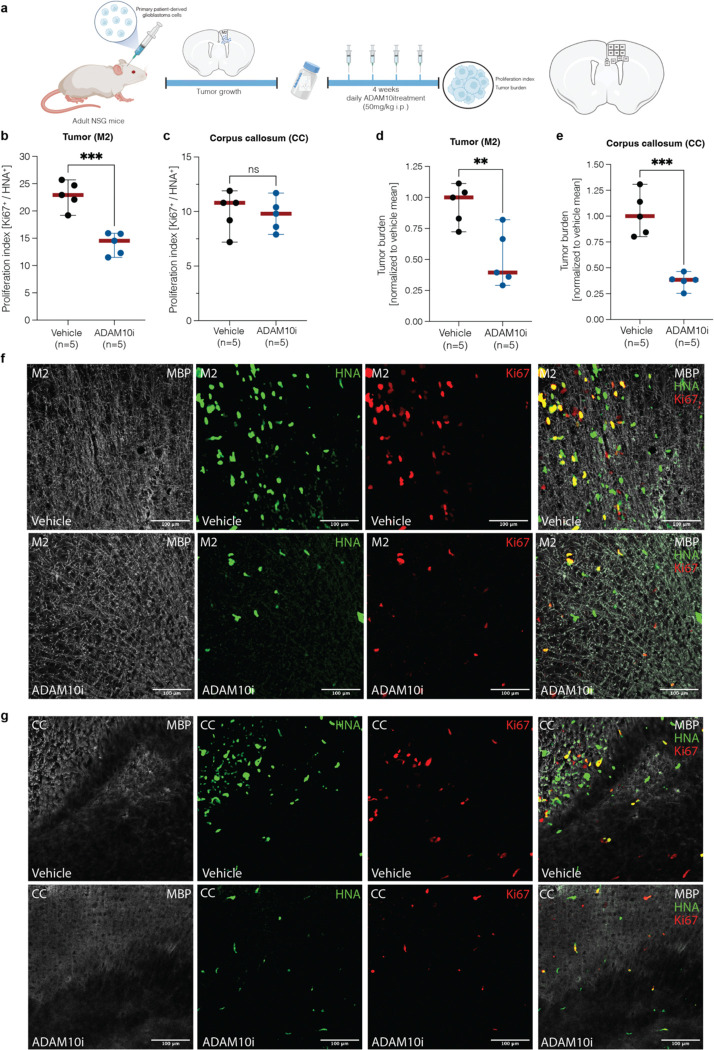
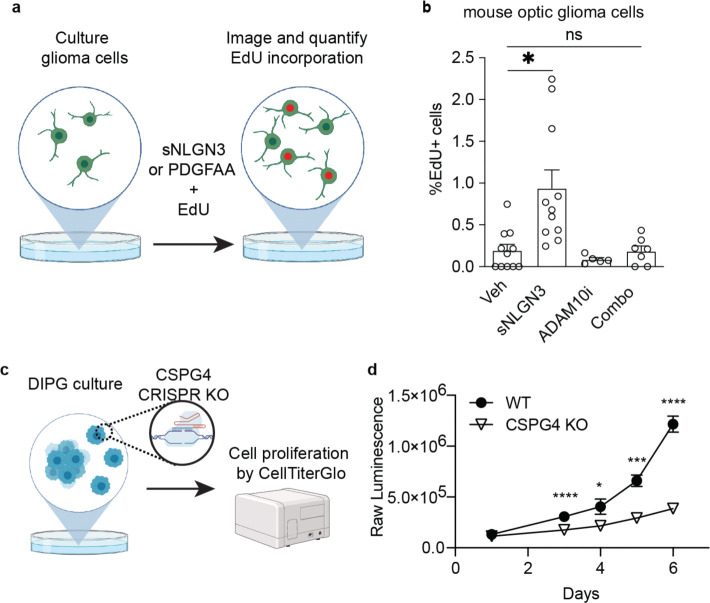
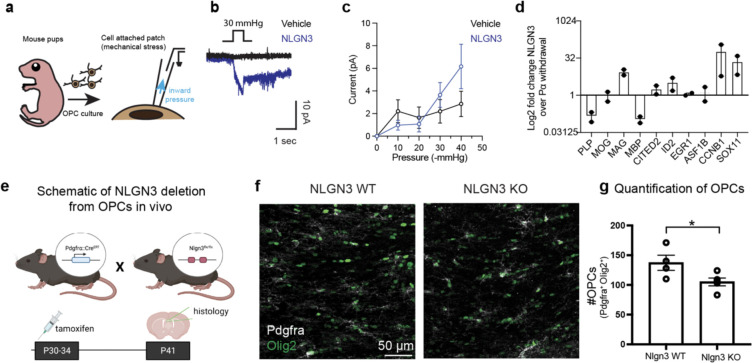
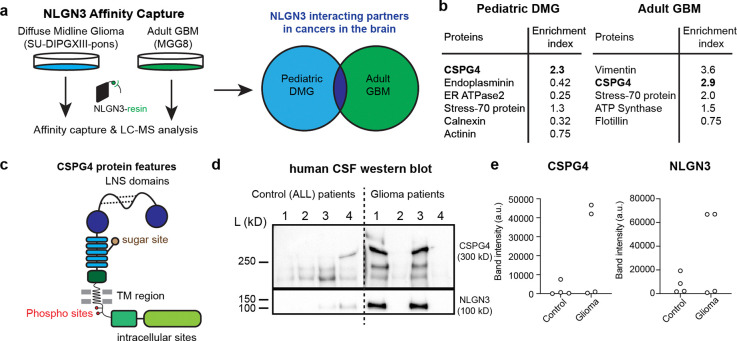
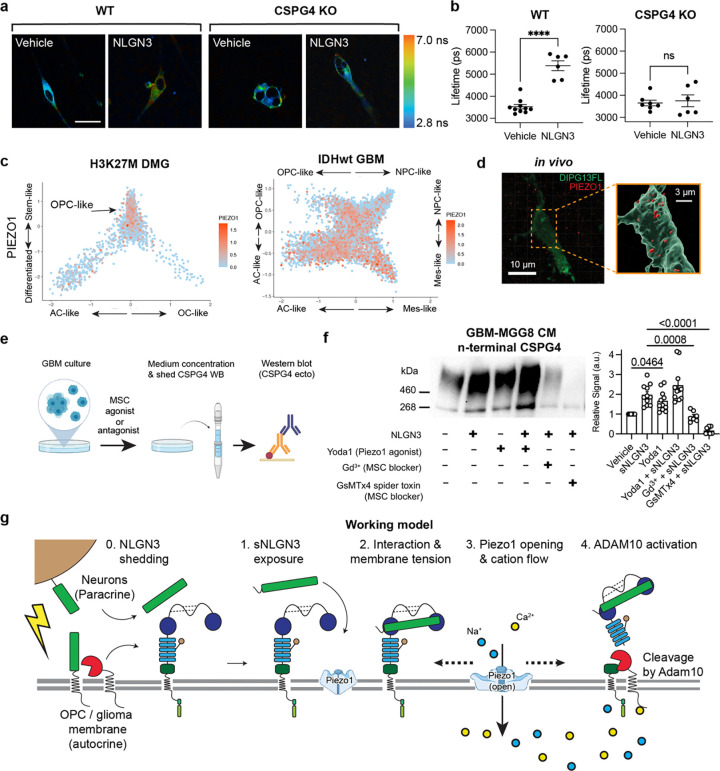
Glioma pathophysiology is robustly regulated by interactions with neurons. Key to these interactions is the role of neuroligin-3 (NLGN3), a synaptic adhesion molecule shed in response to neuronal activity1-5 that functions as a paracrine factor crucial for glioma growth. Here, we elucidate the mechanistic pathway whereby shed NLGN3 interacts with glioma and their normal glial counterpart. NLGN3 interacts with Chondroitin Sulfate Proteoglycan 4 (CSPG4) on both glioma and healthy oligodendrocyte precursor cells (OPCs)6-9, facilitating CSPG4 shedding by ADAM10. NLGN3-CSPG4 interactions and consequent shedding alter membrane tension, thereby activating PIEZO1 mechanosensitive channels and causing membrane depolarization. The NLGN3-CSPG4-PIEZO1 axis maintains OPCs in an undifferentiated, stem-like state and promotes glioma proliferation, underscoring important functional roles for the NLGN3-CSPG4-PIEZO1 axis in both healthy and malignant glial precursors.
Conflict of interest statement
Competing interests M.M. holds equity in MapLight Therapeutics, Stellaromics and holds stock in Cargo Therapeutics.
Figures






References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources