

This is a preprint.
Huntington's disease LIG1 modifier variant increases ligase fidelity and suppresses somatic CAG repeat expansion
- PMID: 40791503
- PMCID: PMC12338604
- DOI: 10.1101/2025.07.15.664798
Huntington's disease LIG1 modifier variant increases ligase fidelity and suppresses somatic CAG repeat expansion
Abstract
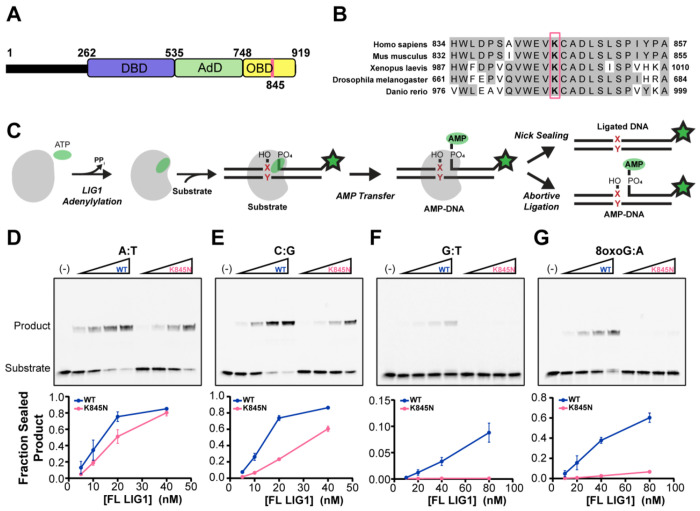
Huntington's disease (HD) is a fatal neurodegenerative disorder caused by inheriting an expanded CAG repeat tract in the huntingtin gene (HTT) that further expands in somatic cells over an individual's lifetime. Genome-wide association studies have provided critical insight into factors that modify the course of disease. These include DNA repair genes that alter the rate of somatic expansion and other genes that do not appear to directly influence this process. One modifier gene is DNA ligase 1 (LIG1), in which a variant specifying a lysine to asparagine substitution (K845N) is associated with a profound (7-8 year) delay in the onset of motor signs. Here, we have taken a multifaceted approach to gain insight into the protective nature of this variant in HD. We demonstrate using in vitro ligase assays and enzyme kinetics that K845N enhances discrimination towards mismatched substrates and increases repair fidelity. Consistent with increased ligation fidelity, K845N confers protection against oxidative stress in cell-based assays. Finally, we demonstrate that the mouse LIG1 K843N orthologue suppresses somatic CAG expansion in HD knock-in mice. Overall, our data provide evidence that altered LIG1 function due to the K845N substitution may contribute to HD clinical delay by slowing somatic expansion in the brain and protecting the genome globally against damage. Significantly, our results provide a mechanistic foundation for considering DNA ligase fidelity as a therapeutic target in HD and potentially in other trinucleotide repeat disorders.
Keywords: Biological Sciences; DNA damage; DNA ligase 1; Genetics; Huntington’s disease; repair fidelity; somatic repeat expansion.
Figures




References
-
- The Huntington’s Disease Collaborative Research Group., A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72, 971–983 (1993). - PubMed
-
- Kennedy L. et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet 12, 3359–3367 (2003). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous