

Organoid-on-a-chip (OrgOC): Advancing cystic fibrosis research
- PMID: 40791795
- PMCID: PMC12336816
- DOI: 10.1016/j.mtbio.2025.102148
Organoid-on-a-chip (OrgOC): Advancing cystic fibrosis research
Abstract
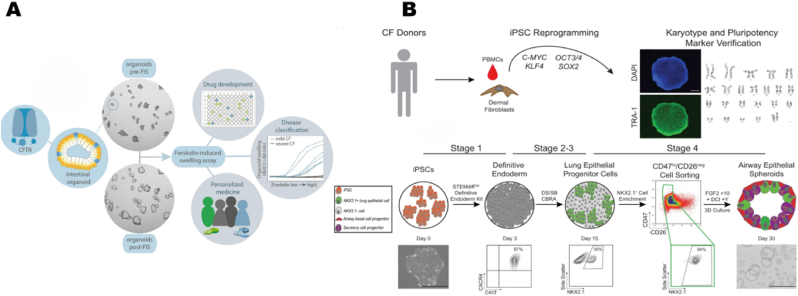
Cystic fibrosis (CF) is an autosomal recessive disorder resulting from impaired anion transport in the epithelium of multiple organs, thereby affecting various physiological functions throughout the body. The heterogeneity of CF complicates drug development, highlighting the growing importance of individualized therapies. CF patient-derived organoid models and organ-on-a-chip (OOC) platforms are promising in vitro models for recapitulating CF pathology, owing to their high simulation fidelity, individualized therapeutic capabilities, cost-effectiveness, and high-throughput screening potential. This review systematically summarizes the technological development pathways of patient-derived organoids and OOC platforms for CF, along with recent advances in their applications to CF-related basic research, and particularly focuses on exploratory studies using organoid-on-a-chip (OrgOC) systems to elucidate CF pathogenesis and assess therapeutic approaches.
Keywords: Cystic fibrosis; Organ-on-a-chip; Organoid; Organoid-on-a-chip.
© 2025 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures






References
-
- Rommens J.M., Iannuzzi M.C., Kerem B.-S., Drumm M.L., Melmer G., Dean M., Rozmahel R., Cole J.L., Kennedy D., Hidaka N., Zsiga M., Buchwald M., Tsui L.-C., Riordan J.R., Collins F.S. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. - DOI - PubMed
-
- Riordan J.R., Rommens J.M., Kerem B.-S., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.-L., Drumm M.L., Iannuzzi M.C., Collins F.S., Tsui L.-C. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
