

Diagnosis and genetic analysis of Gaucher disease in a pediatric case: a case report
- PMID: 40791806
- PMCID: PMC12336233
- DOI: 10.3389/fped.2025.1628525
Diagnosis and genetic analysis of Gaucher disease in a pediatric case: a case report
Abstract
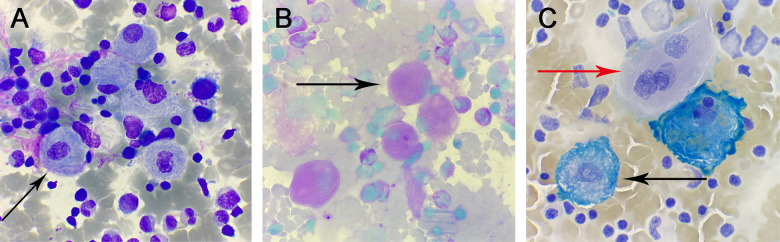
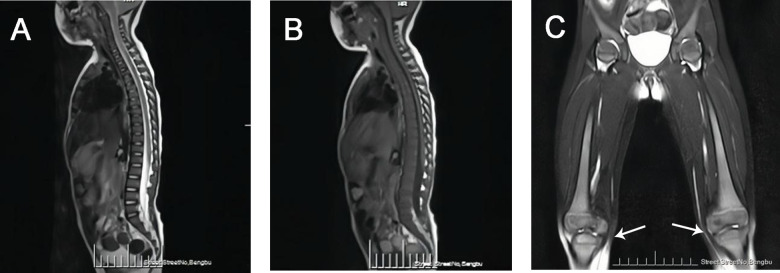
A 2-year-old patient was admitted to our hospital with hepatosplenomegaly as the prominent clinical feature. Peripheral blood analysis during hospitalization revealed trilineage cytopenia. Bone marrow cytology examination demonstrated abundant suspected Gaucher cells. Full-spine MRI exhibited widening of the distal femoral metaphysis with an "Erlenmeyer flask deformity." Subsequent enzymatic and genetic evaluations for Gaucher disease (GD) confirmed reduced β-glucocerebrosidase (GBA) activity, significantly elevated glucosylsphingosine (Lyso-Gb1) levels, and a homozygous missense mutation in the GBA gene c. 1448T>C(p.Leu483Pro). Genetic testing of the parents revealed both were heterozygous carriers of the same mutationc. 1448T>C(p.Leu483Pro), confirming the diagnosis of GD in the child with an autosomal recessive inheritance pattern. GD typically presents in childhood with hepatosplenomegaly, anemia, and thrombocytopenia. Given its rarity and nonspecific clinical manifestations, bone marrow cytology and imaging studies may provide diagnostic clues, but definitive diagnosis requires confirmation through β-glucocerebrosidase activity assays and genetic testing. Enzyme replacement therapy (ERT) is currently the primary treatment modality. The child is receiving regular intravenous infusions of imiglucerase at our hospital.
Keywords: Gaucher disease; Mendelian inheritance; genetic testing; rare disease; β-glucocerebrosidase.
© 2025 Ma, Wu, Feng, Sang, Duan, Li and Zhang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous