

Hyperactive PLCG1 induces cell-autonomous and bystander T cell activation and drug resistance
- PMID: 40796680
- PMCID: PMC12457681
- DOI: 10.1038/s44319-025-00546-x
Hyperactive PLCG1 induces cell-autonomous and bystander T cell activation and drug resistance
Abstract
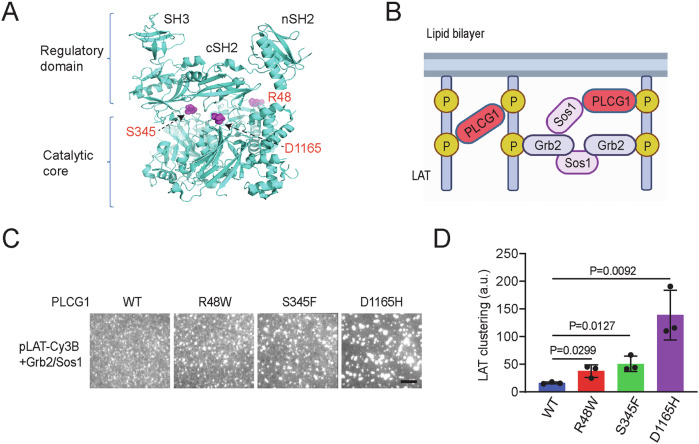
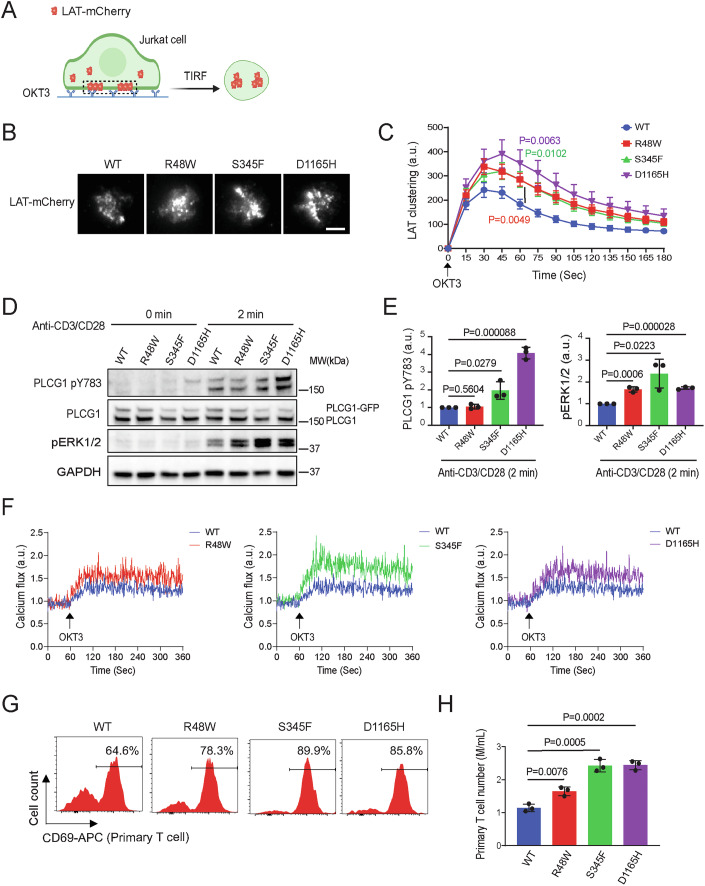
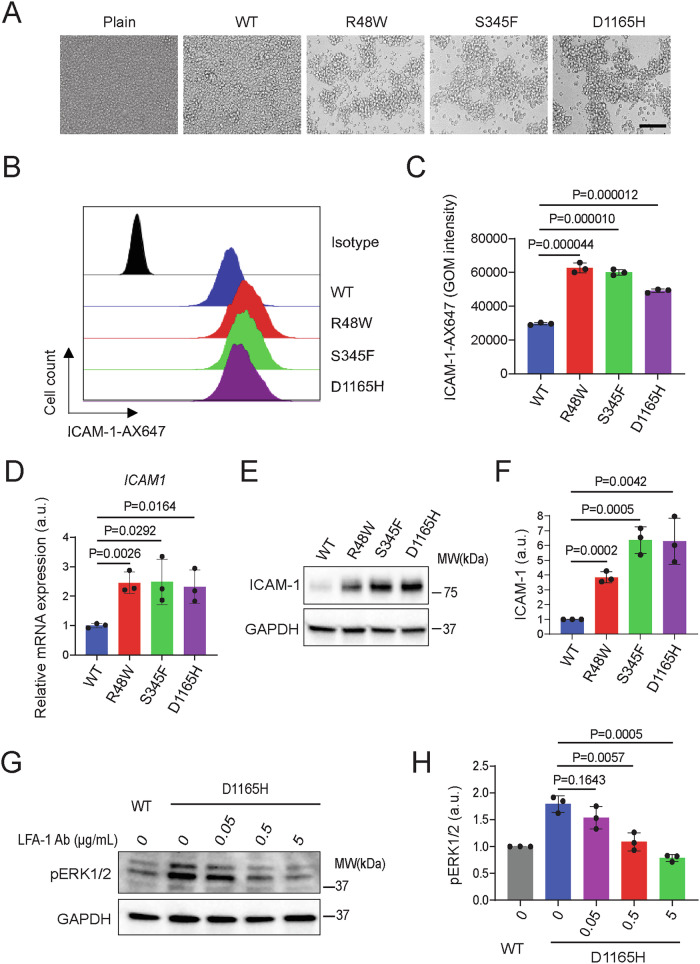
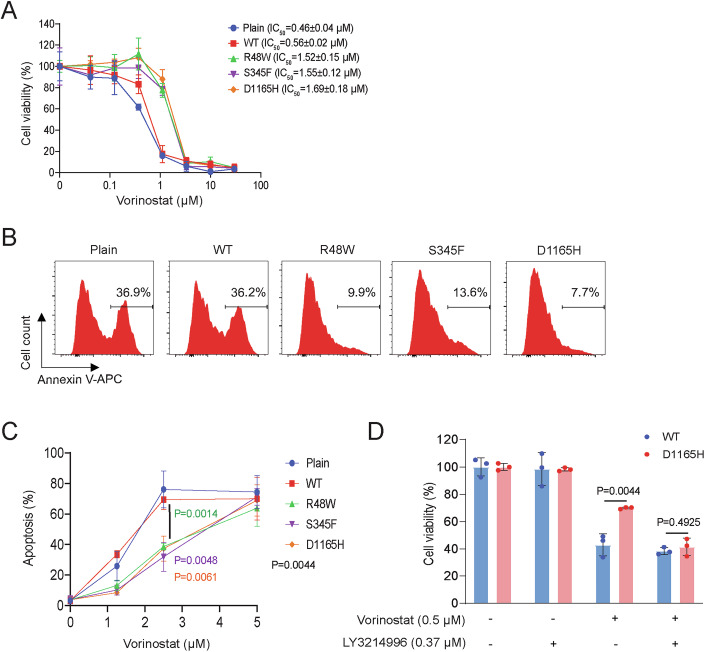
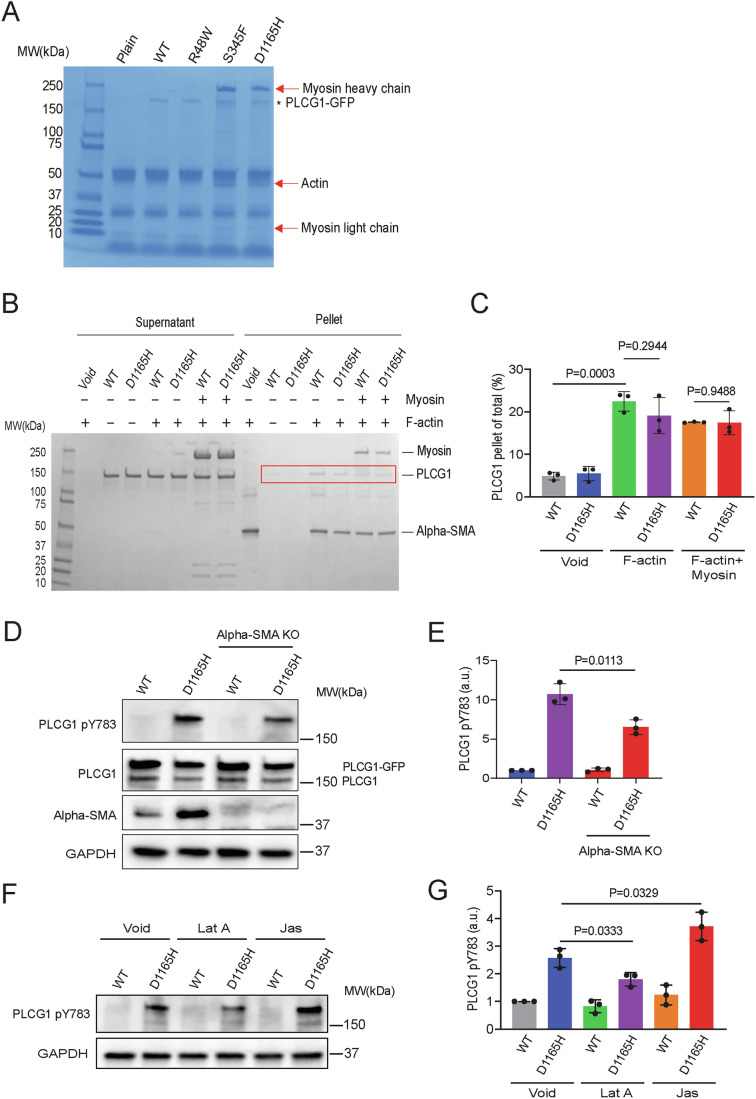
Phospholipase C gamma 1 (PLCG1) has been identified as the most frequently mutated gene in adult T-cell leukemia/lymphoma, suggesting a critical function of PLCG1 in driving T cell activation. However, it remains unclear how these mutations regulate T cell physiology and pathology. Here, we investigate three common leukemia/lymphoma-associated mutations (R48W, S345F, and D1165H). We discover that these mutations induce hyperactive T cell signaling and cause pro-survival phenotypes. PLCG1 mutants enhance LAT condensation, calcium influx, and ERK activation. They also promote T cell proliferation, upregulate cell adhesion molecules, induce cell aggregation, and confer resistance to Vorinostat, an FDA-approved drug for cutaneous T-cell lymphoma. The resistance depends on ERK signaling and can be reversed with an ERK inhibitor. Interestingly, PLCG1 mutants also induce bystander drug resistance in nearby cells expressing wild-type PLCG1. Mechanistically, alpha smooth muscle actin, which is specifically induced by PLCG1 mutants, directly binds PLCG1 to promote its activation. These results demonstrate that hyperactive PLCG1 promotes T cell survival and drug resistance by inducing non-canonical signaling.
Keywords: Actin; Condensation; ERK; PLCG1; T Cell.
© 2025. The Author(s).
Conflict of interest statement
Disclosure and competing interests statement. The authors declare no competing interests.
Figures








Update of
-
Hyperactive PLCG1 drives non-canonical signaling to promote cell survival.bioRxiv [Preprint]. 2024 Dec 18:2024.12.17.628879. doi: 10.1101/2024.12.17.628879. bioRxiv. 2024. Update in: EMBO Rep. 2025 Sep;26(18):4563-4586. doi: 10.1038/s44319-025-00546-x. PMID: 39763946 Free PMC article. Updated. Preprint.
References
-
- Ananthanarayanan B, Das S, Rhee SG, Murray D, Cho W (2002) Membrane targeting of C2 domains of phospholipase C-delta isoforms. J Biol Chem 277:3568–3575 - PubMed
MeSH terms
Substances
Grants and funding
- CA016086/HHS | NIH | National Institute of General Medical Sciences (NIGMS)
- CA121974/Yale Cancer Center | Yale SPORE in Lung Cancer
- R35 GM149299/GM/NIGMS NIH HHS/United States
- AG083424/HHS | NIH | National Institute of General Medical Sciences (NIGMS)
- RGY0088/2021/Human Frontier Science Program (HFSP)
- P30 CA016086/CA/NCI NIH HHS/United States
- R21 CA294038/CA/NCI NIH HHS/United States
- R21 CA286364/CA/NCI NIH HHS/United States
- GM149299/HHS | NIH | National Institute of General Medical Sciences (NIGMS)
- P50 CA121974/CA/NCI NIH HHS/United States
- CA258993/HHS | NIH | National Institute of General Medical Sciences (NIGMS)
- P30 DK034989/DK/NIDDK NIH HHS/United States
- 22YIC53/Rally Foundation (Rally Foundation, Inc.)
- CRI3516/Cancer Research Institute (CRI)
- JP21H05051/MEXT | Japan Society for the Promotion of Science (JSPS)
- GM138299/HHS | NIH | National Institute of General Medical Sciences (NIGMS)
- DK034989/Yale | YSM | Yale Liver Center
- CA286364/HHS | NIH | NCI | Division of Cancer Prevention, National Cancer Institute (DCP, NCI)
- EB037112/HHS | NIH | National Institute of General Medical Sciences (NIGMS)
- CA294038/HHS | NIH | National Institute of General Medical Sciences (NIGMS)
- R35 GM138299/GM/NIGMS NIH HHS/United States
- R01 EB037112/EB/NIBIB NIH HHS/United States
- R01 CA258993/CA/NCI NIH HHS/United States
- R56 AG083424/AG/NIA NIH HHS/United States
- 135926/American Cancer Society (ACS)
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
