

Mutations of short tandem repeats explain abundant trait heritability in Arabidopsis
- PMID: 40796852
- PMCID: PMC12341274
- DOI: 10.1186/s13059-025-03720-5
Mutations of short tandem repeats explain abundant trait heritability in Arabidopsis
Abstract
Background: Short tandem repeat (STR) mutations are major drivers of genetic variation and deeply influence phenotypic diversity and evolution, they are often overlooked despite their significant effects.
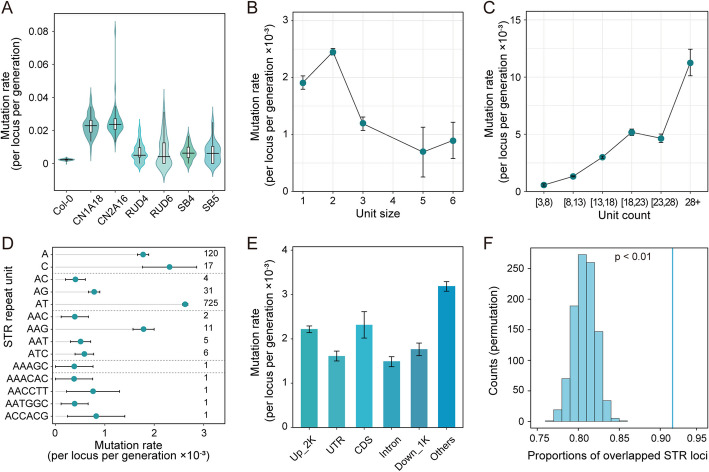
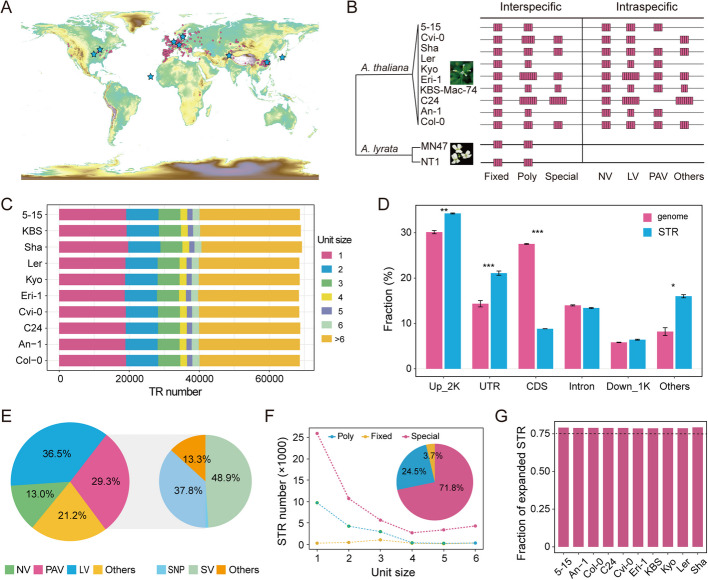
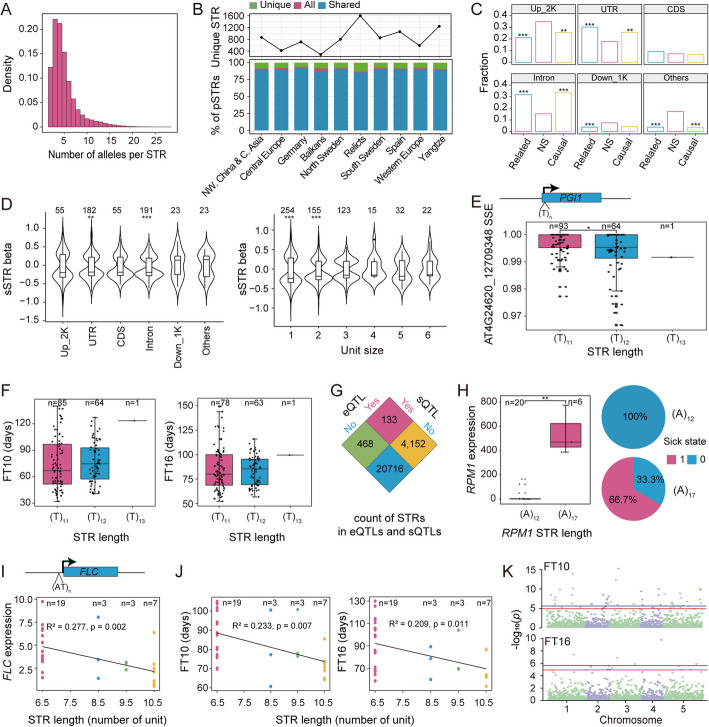
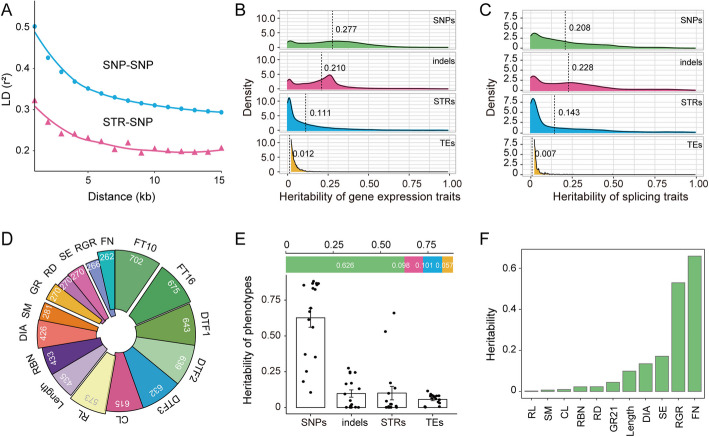
Results: Here, we leverage mutation accumulation lines descended from Col-0 accession of Arabidopsis thaliana to assess the variation in the repeat length of STRs (STR mutation rate). We find that STR mutation rate far exceeds single nucleotide polymorphisms rates. Interspecific comparison between A. thaliana and Arabidopsis lyrata reveals rapid STR turnover, with the most majority of the loci occurring only in A. thaliana. Intraspecific comparison of ten assembled A. thaliana genomes reveals that 29.3% of STRs display presence/absence variations, 36.5% show length variation, 21.2% have both types of variations, while only a small proportion have no variation. By association analysis, we find several STRs are associated with diverse phenotypes. Further analysis based on RNA-seq dataset from 413 accessions, we identify 3,871 expression-associated STRs and 651 splicing-associated STRs, of which over one thousand co-localized with known signals for diverse traits detected by genome-wide association studies. Notably, based on analysis of the expression levels of 24,175 genes and splice site strength values of 12,784 splice sites, as well as 16 phenotypes of natural A. thaliana populations, we determine the similar average heritability of these three trait sets explained by STR variation.
Conclusions: Our results reveal the evolutionary dynamics of STRs, and highlight the importance of STR variation as an important contributor to missing heritability in regulating complex traits.
Keywords: Arabidopsis thaliana; Evolution; Missing heritability; Mutation rate; Natural variation; Short tandem repeats.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
