

Familial spinocerebellar ataxia type 3: A case report of multi-generational presentation
- PMID: 40797466
- PMCID: PMC12338283
- DOI: 10.1097/MD.0000000000043812
Familial spinocerebellar ataxia type 3: A case report of multi-generational presentation
Abstract
Rationale: Spinocerebellar ataxia type 3 (SCA3), also known as Machado-Joseph disease, a rare autosomal dominant neurodegenerative disorder caused by cytosine-adenine-guanine repeat expansions in ATXN3, lacks effective therapies. This case report highlights the clinical and genetic features of a family with 5 affected members to emphasize the challenges in diagnosis, management, and the need for targeted therapies.
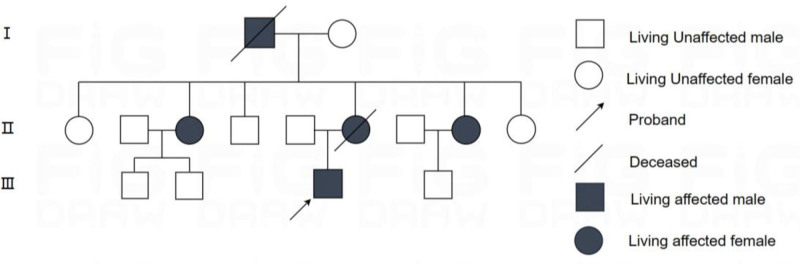
Patient concerns: A 33-year-old male proband presented with progressive gait instability, limb incoordination, and dysarthria for more than 2 years. The symptoms worsened in cold weather and were accompanied by head swelling and muscle weakness. The patient reported a strong family history of similar neurological symptoms across the 3 generations.
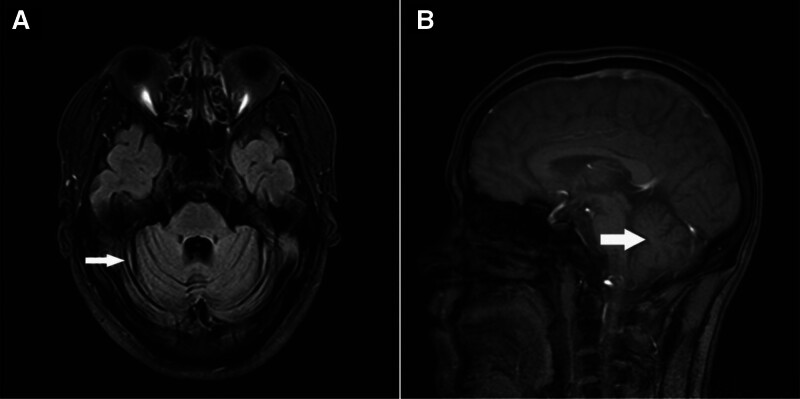
Diagnosis: Clinical evaluation revealed cerebellar ataxia, a wide-based gait, and impaired coordination. Brain MRI revealed bilateral cerebellar atrophy. Genetic testing confirmed the presence of a pathogenic ATXN3 allele with 78 cytosine-adenine-guanine repeats (normal range: ≤49), which is consistent with SCA3. Familial genetic analysis identified identical mutations in 4 additional relatives.
Interventions: Supportive treatment included improvement in circulation, neuroprotective agents, and symptomatic management. No disease-modifying therapies were administered, owing to their limited availability.
Outcomes: The patient's condition did not improve during hospitalization, reflecting the progressive nature of the SCA3. Similar outcomes were observed in affected family members.
Lessons: Early genetic testing is critical for a definitive diagnosis, especially in familial cases. The lack of effective therapies underscores the urgency of clinical trials that target polyglutamine toxicity. Multidisciplinary care and patient education are essential for the management of this debilitating disease.
Keywords: MJD; SCA3; family; gene mutation; spinocerebellar ataxia type 3.
Copyright © 2025 the Author(s). Published by Wolters Kluwer Health, Inc.
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Figures
Similar articles
-
Clinical Characteristics of Spinocerebellar Ataxia Type 3 in Uruguay.Cerebellum. 2025 Apr 28;24(4):89. doi: 10.1007/s12311-025-01839-6. Cerebellum. 2025. PMID: 40289053
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Spinal Muscular Atrophy.2000 Feb 24 [updated 2024 Sep 19]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2000 Feb 24 [updated 2024 Sep 19]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301526 Free Books & Documents. Review.
-
Friedreich Ataxia.1998 Dec 18 [updated 2025 Jun 26]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 1998 Dec 18 [updated 2025 Jun 26]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301458 Free Books & Documents. Review.
-
PTS-Related Tetrahydrobiopterin Deficiency (PTPSD).2025 Jul 10. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2025 Jul 10. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 40638773 Free Books & Documents. Review.
References
-
- Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–94. - PubMed
-
- Jazurek-Ciesiolka M, Ciesiolka A, Komur AA, Urbanek-Trzeciak MO, Krzyzosiak WJ, Fiszer A. RAN translation of the expanded CAG repeats in the SCA3 disease context. J Mol Biol. 2020;432:166699. - PubMed
-
- Raposo M, Hübener-Schmid J, Tagett R, et al. ; European Spinocerebellar ataxia type 3/Machado-Joseph disease Initiative (ESMI) study group. Blood and cerebellar abundance of ATXN3 splice variants in spinocerebellar ataxia type 3/Machado-Joseph disease. Neurobiol Dis. 2024;193:106456. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
