

Estimating protein complex model accuracy using graph transformers and pairwise similarity graphs
- PMID: 40799498
- PMCID: PMC12342149
- DOI: 10.1093/bioadv/vbaf180
Estimating protein complex model accuracy using graph transformers and pairwise similarity graphs
Abstract
Motivation: Estimation of protein complex structure accuracy is essential for effective structural model selection in structural biology applications such as protein function analysis and drug design. Despite the success of structure prediction methods such as AlphaFold2 and AlphaFold3, selecting top-quality structural models from large model pools remains challenging.
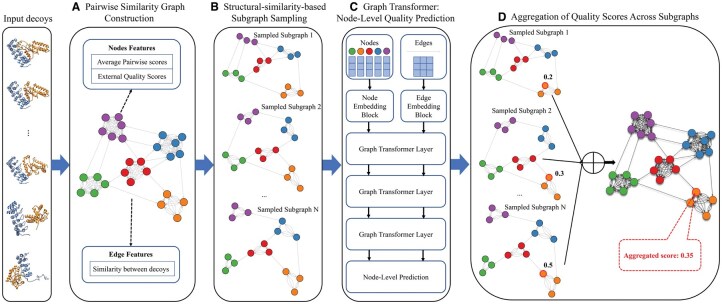
Results: We present GATE, a novel method that uses graph transformers on pairwise model similarity graphs to predict the quality (accuracy) of complex structural models. By integrating single-model and multimodel quality features, GATE captures intrinsic model characteristics and intermodel geometric similarities to make robust predictions. On the dataset of the 15th Critical Assessment of Protein Structure Prediction (CASP15), GATE achieved the highest Pearson's correlation (0.748) and the lowest ranking loss (0.1191) compared with existing methods. In the blind CASP16 experiment, GATE ranked fifth based on the sum of z-scores, with a Pearson's correlation of 0.7076 (first), a Spearman's correlation of 0.4514 (fourth), a ranking loss of 0.1221 (third), and an area under the curve score of 0.6680 (third) on per-target TM-score-based metrics. Additionally, GATE also performed consistently on large in-house datasets generated by extensive AlphaFold-based sampling with MULTICOM4, confirming its robustness and practical applicability in real-world model selection scenarios.
Availability and implementation: GATE is available at https://github.com/BioinfoMachineLearning/GATE.
© The Author(s) 2025. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures
Update of
-
Estimating Protein Complex Model Accuracy Using Graph Transformers and Pairwise Similarity Graphs.bioRxiv [Preprint]. 2025 Feb 23:2025.02.04.636562. doi: 10.1101/2025.02.04.636562. bioRxiv. 2025. Update in: Bioinform Adv. 2025 Jul 29;5(1):vbaf180. doi: 10.1093/bioadv/vbaf180. PMID: 39975041 Free PMC article. Updated. Preprint.
Similar articles
-
Boosting AlphaFold Protein Tertiary Structure Prediction through MSA Engineering and Extensive Model Sampling and Ranking in CASP16.bioRxiv [Preprint]. 2025 Jun 9:2025.06.06.658338. doi: 10.1101/2025.06.06.658338. bioRxiv. 2025. PMID: 40661500 Free PMC article. Preprint.
-
Estimating Protein Complex Model Accuracy Using Graph Transformers and Pairwise Similarity Graphs.bioRxiv [Preprint]. 2025 Feb 23:2025.02.04.636562. doi: 10.1101/2025.02.04.636562. bioRxiv. 2025. Update in: Bioinform Adv. 2025 Jul 29;5(1):vbaf180. doi: 10.1093/bioadv/vbaf180. PMID: 39975041 Free PMC article. Updated. Preprint.
-
Assessment of Protein Complex Predictions in CASP16: Are we making progress?bioRxiv [Preprint]. 2025 May 30:2025.05.29.656875. doi: 10.1101/2025.05.29.656875. bioRxiv. 2025. PMID: 40501681 Free PMC article. Preprint.
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
-
Artificial intelligence for diagnosing exudative age-related macular degeneration.Cochrane Database Syst Rev. 2024 Oct 17;10(10):CD015522. doi: 10.1002/14651858.CD015522.pub2. Cochrane Database Syst Rev. 2024. PMID: 39417312
Cited by
-
Boosting AlphaFold Protein Tertiary Structure Prediction through MSA Engineering and Extensive Model Sampling and Ranking in CASP16.bioRxiv [Preprint]. 2025 Jun 9:2025.06.06.658338. doi: 10.1101/2025.06.06.658338. bioRxiv. 2025. PMID: 40661500 Free PMC article. Preprint.
-
Boosting AlphaFold Protein Tertiary Structure Prediction through MSA Engineering and Extensive Model Sampling and Ranking in CASP16.Res Sq [Preprint]. 2025 Jun 20:rs.3.rs-6845168. doi: 10.21203/rs.3.rs-6845168/v1. Res Sq. 2025. PMID: 40585263 Free PMC article. Preprint.
-
Improving AlphaFold2- and AlphaFold3-Based Protein Complex Structure Prediction With MULTICOM4 in CASP16.Proteins. 2025 Jun 2:10.1002/prot.26850. doi: 10.1002/prot.26850. Online ahead of print. Proteins. 2025. PMID: 40452318
References
Grants and funding
LinkOut - more resources
Full Text Sources
