

Reactive Oxygen Species: A Double-Edged Sword in the Modulation of Cancer Signaling Pathway Dynamics
- PMID: 40801639
- PMCID: PMC12346646
- DOI: 10.3390/cells14151207
Reactive Oxygen Species: A Double-Edged Sword in the Modulation of Cancer Signaling Pathway Dynamics
Abstract
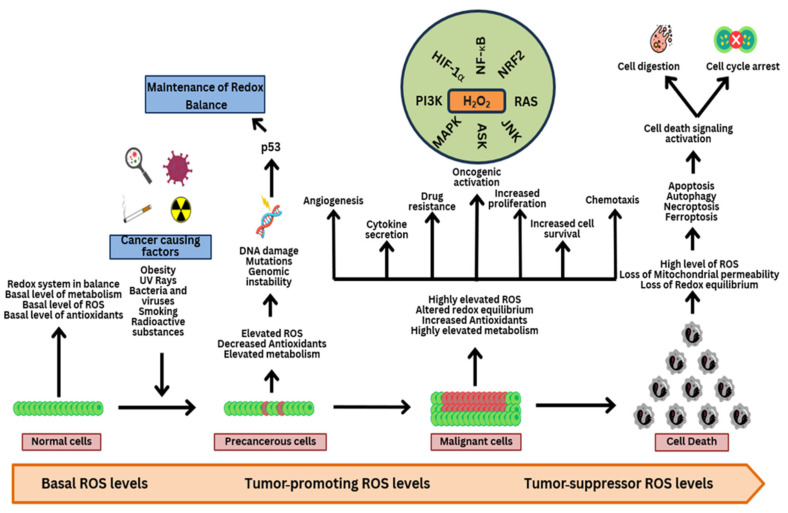
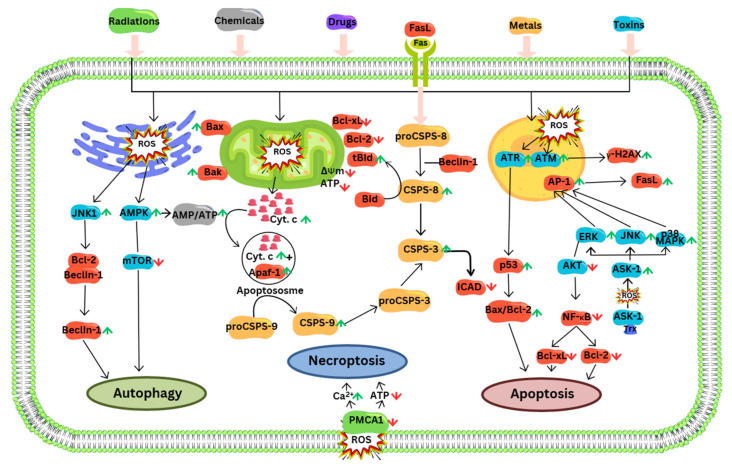
Reactive oxygen species (ROS) are often seen solely as harmful byproducts of oxidative metabolism, yet evidence reveals their paradoxical roles in both promoting and inhibiting cancer progression. Despite advances, precise context-dependent mechanisms by which ROS modulate oncogenic signaling, therapeutic response, and tumor microenvironment dynamics remain unclear. Specifically, the spatial and temporal aspects of ROS regulation (i.e., the distinct effects of mitochondrial versus cytosolic ROS on the PI3K/Akt and NF-κB pathways, and the differential cellular outcomes driven by acute versus chronic ROS exposure) have been underexplored. Additionally, the specific contributions of ROS-generating enzymes, like NOX isoforms and xanthine oxidase, to tumor microenvironment remodeling and immune modulation remain poorly understood. This review synthesizes current findings with a focus on these critical gaps, offering novel mechanistic insights into the dualistic nature of ROS in cancer biology. By systematically integrating data on ROS source-specific functions and redox-sensitive signaling pathways, the complex interplay between ROS concentration, localization, and persistence is elucidated, revealing how these factors dictate the paradoxical support of tumor progression or induction of cancer cell death. Particular attention is given to antioxidant mechanisms, including NRF2-mediated responses, that may undermine the efficacy of ROS-targeted therapies. Recent breakthroughs in redox biosensors (i.e., redox-sensitive fluorescent proteins, HyPer variants, and peroxiredoxin-FRET constructs) enable precise, real-time ROS imaging across subcellular compartments. Translational advances, including redox-modulating drugs and synthetic lethality strategies targeting glutathione or NADPH dependencies, further highlight actionable vulnerabilities. This refined understanding advances the field by highlighting context-specific vulnerabilities in tumor redox biology and guiding more precise therapeutic strategies. Continued research on redox-regulated signaling and its interplay with inflammation and therapy resistance is essential to unravel ROS dynamics in tumors and develop targeted, context-specific interventions harnessing their dual roles.
Keywords: ROS; antioxidants; cancer; oncogenic signaling circuits; oxidative stress mediators; programmed cell death.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures


Similar articles
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
NADPH oxidases: redox regulation of cell homeostasis and disease.Physiol Rev. 2025 Jul 1;105(3):1291-1428. doi: 10.1152/physrev.00034.2023. Epub 2025 Jan 15. Physiol Rev. 2025. PMID: 39814410 Free PMC article. Review.
-
Redox-signalling and Redox Biomarkers in Cardiovascular Health and Disease.Cardiovasc Hematol Agents Med Chem. 2025;23(2):99-111. doi: 10.2174/0118715257282030240130095754. Cardiovasc Hematol Agents Med Chem. 2025. PMID: 38549520 Review.
-
The Redox Revolution in Brain Medicine: Targeting Oxidative Stress with AI, Multi-Omics and Mitochondrial Therapies for the Precision Eradication of Neurodegeneration.Int J Mol Sci. 2025 Aug 3;26(15):7498. doi: 10.3390/ijms26157498. Int J Mol Sci. 2025. PMID: 40806624 Free PMC article. Review.
-
Redox signaling in innate immunity and inflammation: focus on macrophages and neutrophils.Free Radic Biol Med. 2025 Sep;237:427-454. doi: 10.1016/j.freeradbiomed.2025.06.006. Epub 2025 Jun 6. Free Radic Biol Med. 2025. PMID: 40484207 Review.
References
-
- Krieghoff-Henning E., Folkerts J., Penzkofer A., Weg-Remers S. Cancer—An Overview. Med. Monatsschr. Pharm. 2017;40:48–54. - PubMed
-
- Behl T., Kumar A., Vishakha, Sehgal A., Singh S., Sharma N., Yadav S., Rashid S., Ali N., Ahmed A.S., et al. Understanding the Mechanistic Pathways and Clinical Aspects Associated with Protein and Gene Based Biomarkers in Breast Cancer. Int. J. Biol. Macromol. 2023;253:126595. doi: 10.1016/j.ijbiomac.2023.126595. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
