

Microtubule mechanotransduction refines cytomegalovirus interactions with and remodeling of host chromatin
- PMID: 40804345
- PMCID: PMC12350610
- DOI: 10.1038/s41467-025-62921-5
Microtubule mechanotransduction refines cytomegalovirus interactions with and remodeling of host chromatin
Abstract
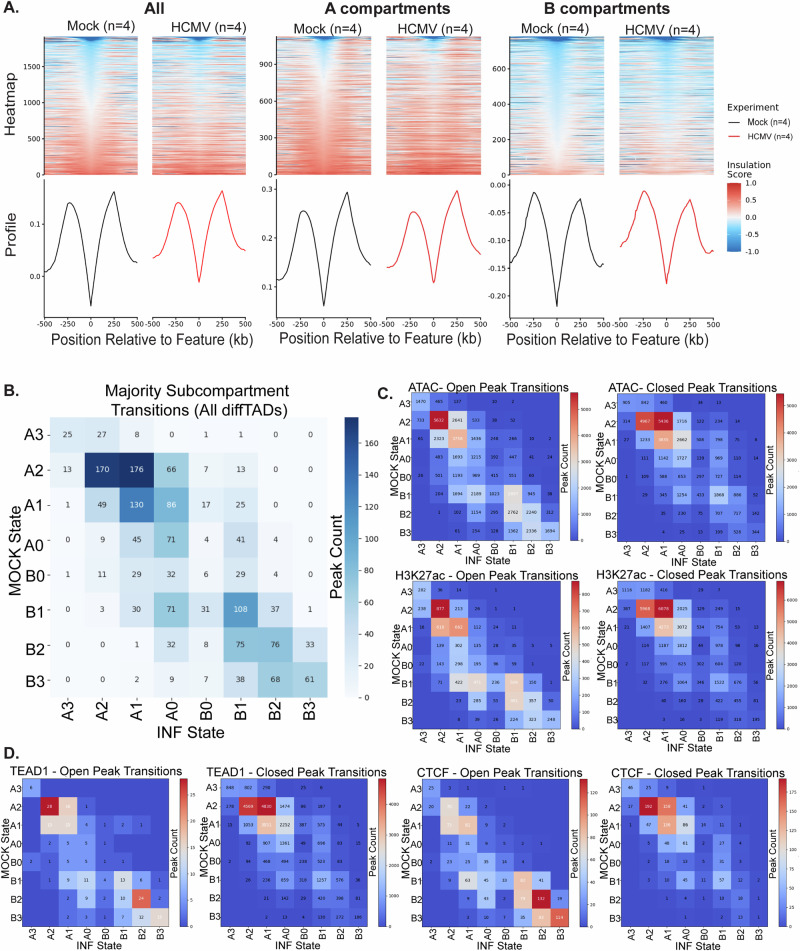
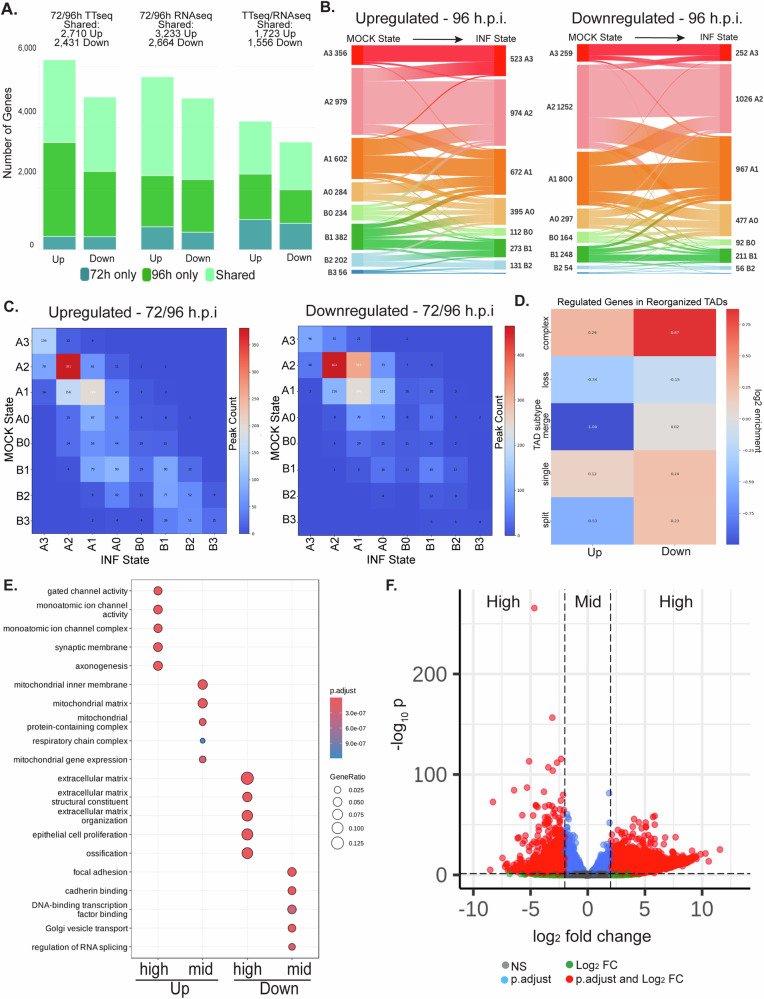
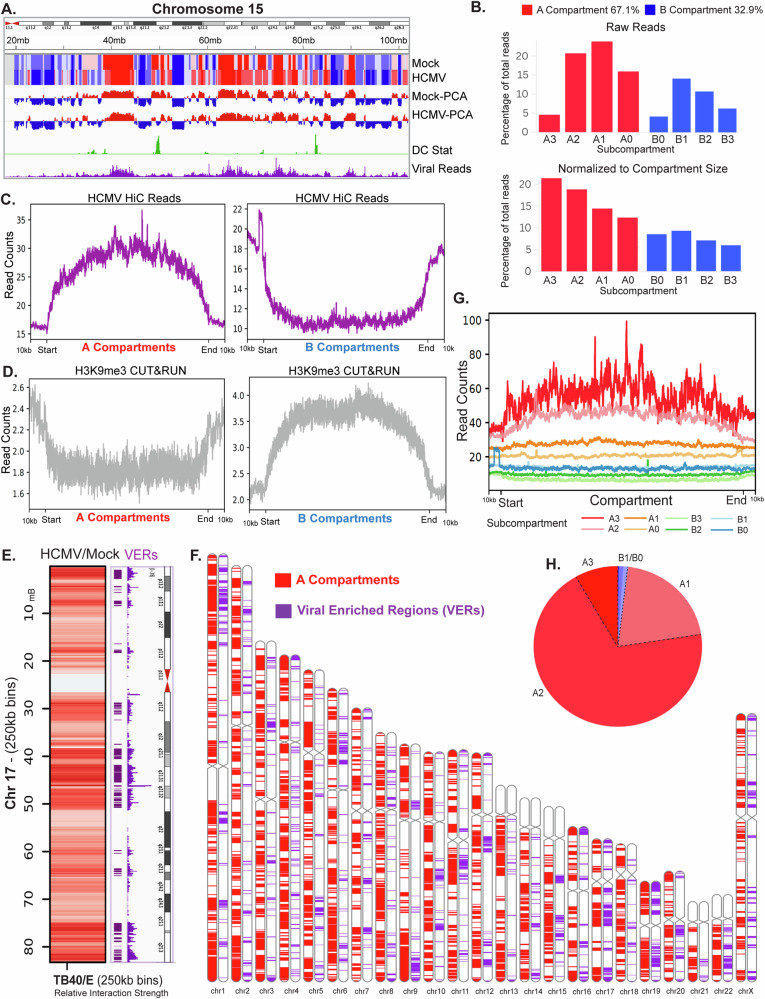
Human cytomegalovirus extensively alters nuclear organization and the cellular transcriptome, yet understanding of these genome-wide events remains relatively limited. Here, chromatin conformation capture (Hi-C) revealed how cytomegalovirus alters chromosome organization at both large- and small-scales. Nascent transcriptomics further revealed how transcriptional changes correlate with genomic reorganization, while also uncovering infection-induced transcriptional dysregulation that contributes to the induction of neuronal gene signatures in infected fibroblasts. Combining Hi-C and Cleavage Under Targets & Release Using Nuclease (CUT&RUN) we find that viral genomes preferentially localize to highly euchromatic compartments, further dysregulating transcription of host genes. Finally, RNAi-mediated depletion of two key effectors of microtubule-based forces that are exerted on the nucleus provides insights into their diverging roles in regulating compartment-scale contacts and viral genomic interactions with host chromatin. Combined, we reveal the extent to which HCMV interacts with and alters host chromatin and transcription, and the influence of microtubule mechanotransduction on these processes.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures




Similar articles
-
Dynamic interactions of retroviral Gag condensates with nascent viral RNA at transcriptional burst sites: implications for genomic RNA packaging.bioRxiv [Preprint]. 2025 Jun 2:2025.01.11.632546. doi: 10.1101/2025.01.11.632546. bioRxiv. 2025. PMID: 39829876 Free PMC article. Preprint.
-
Adenovirus Remodeling of the Host Proteome and Host Factors Associated with Viral Genomes.mSystems. 2021 Aug 31;6(4):101128msystems0046821. doi: 10.1128/mSystems.00468-21. Epub 2021 Aug 31. mSystems. 2021. PMID: 34463575 Free PMC article.
-
Human cytomegalovirus induces neuronal gene expression through IE1 for viral maturation.Nat Commun. 2025 Aug 8;16(1):7316. doi: 10.1038/s41467-025-61915-7. Nat Commun. 2025. PMID: 40781068 Free PMC article.
-
Measures implemented in the school setting to contain the COVID-19 pandemic.Cochrane Database Syst Rev. 2022 Jan 17;1(1):CD015029. doi: 10.1002/14651858.CD015029. Cochrane Database Syst Rev. 2022. Update in: Cochrane Database Syst Rev. 2024 May 2;5:CD015029. doi: 10.1002/14651858.CD015029.pub2. PMID: 35037252 Free PMC article. Updated.
-
From gene to mechanics: a comprehensive insight into the mechanobiology of LMNA mutations in cardiomyopathy.Cell Commun Signal. 2024 Mar 27;22(1):197. doi: 10.1186/s12964-024-01546-5. Cell Commun Signal. 2024. PMID: 38539233 Free PMC article. Review.
References
Grants and funding
LinkOut - more resources
Full Text Sources
