

Are Hippocampal Hypoperfusion and ATP Depletion Prime Movers in the Genesis of Alzheimer's Disease? A Review of Recent Pertinent Observations from Molecular Biology
- PMID: 40806461
- PMCID: PMC12347683
- DOI: 10.3390/ijms26157328
Are Hippocampal Hypoperfusion and ATP Depletion Prime Movers in the Genesis of Alzheimer's Disease? A Review of Recent Pertinent Observations from Molecular Biology
Abstract
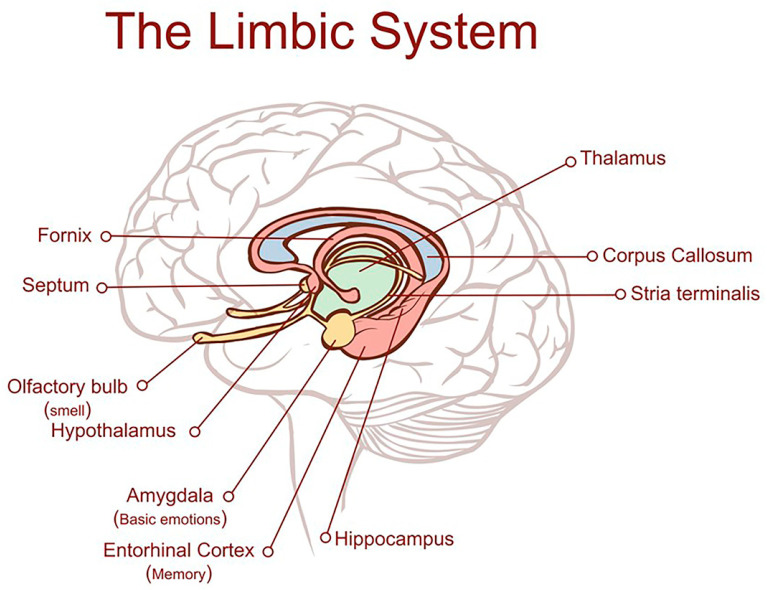
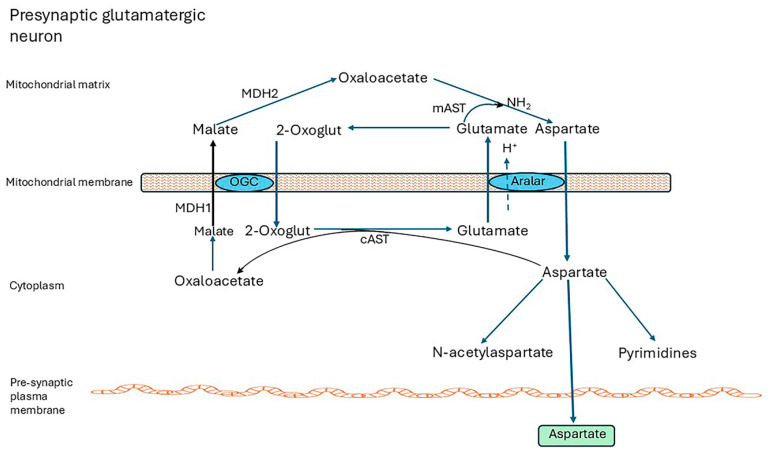
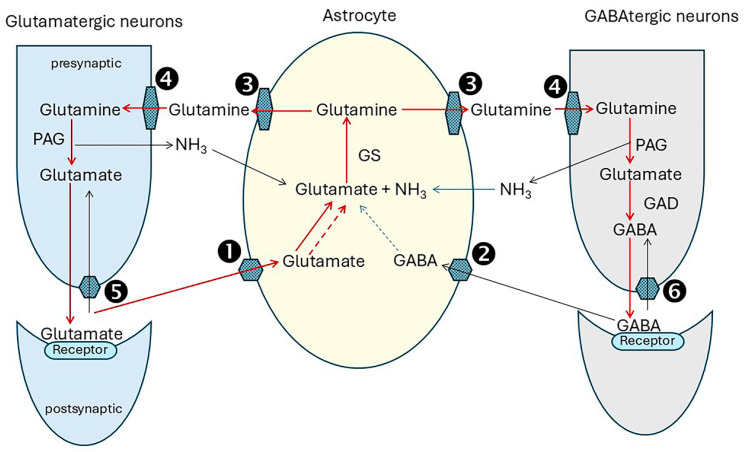
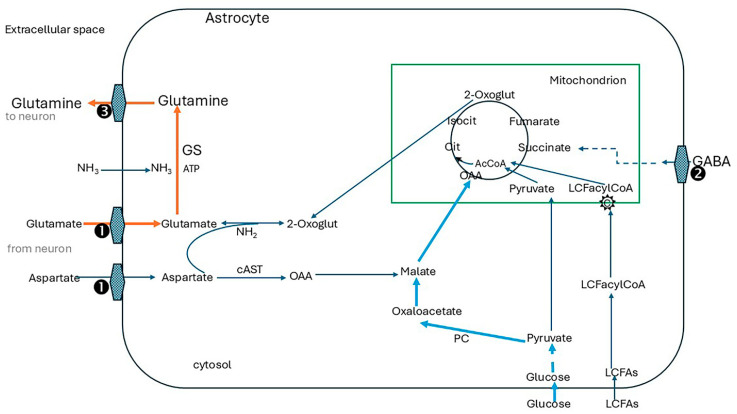
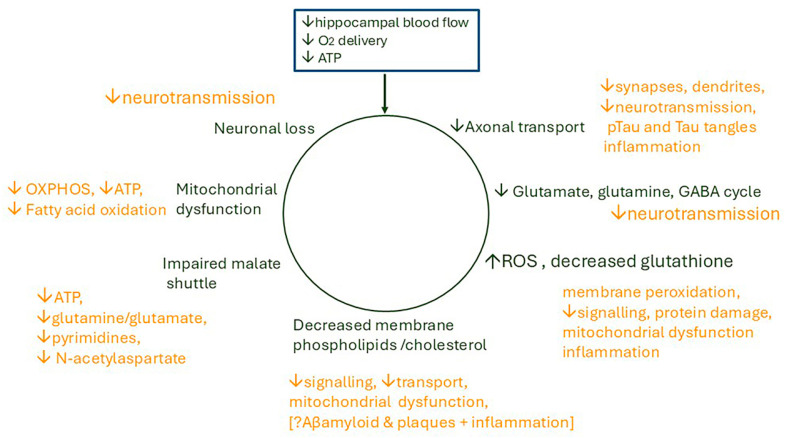
Alzheimer's dementia (AD) is a disease of the ageing brain. It begins in the hippocampal region with the epicentre in the entorhinal cortex, then gradually extends into adjacent brain areas involved in memory and cognition. The events which initiate the damage are unknown and under intense investigation. Localization to the hippocampus can now be explained by anatomical features of the blood vessels supplying this region. Blood supply and hence oxygen delivery to the area are jeopardized by poor flow through narrowed arteries. In genomic and metabolomic studies, the respiratory chain and mitochondrial pathways which generate ATP were leading pathways associated with AD. This review explores the notion that ATP depletion resulting from hippocampal hypoperfusion has a prime role in initiating damage. Sections cover sensing of ATP depletion and protective responses, vulnerable processes with very heavy ATP consumption (the malate shuttle, the glutamate/glutamine/GABA (γ-aminobutyric acid) cycle, and axonal transport), phospholipid disturbances and peroxidation by reactive oxygen species, hippocampal perfusion and the effects of hypertension, chronic hypoxia, and arterial vasospasm, and an overview of recent relevant genomic studies. The findings demonstrate strong scientific arguments for the proposal with increasing supportive evidence. These lines of enquiry should be pursued.
Keywords: ATP biosensors; axonal transport; cerebral arterial perfusion; glutamate/GABA/glutamine cycle; malate aspartate shuttle; membrane phospholipids; mitochondrial-derived peptides; vasospasm.
Conflict of interest statement
The author declares no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
