

Lithuanian Study on COL4A3 and COL4A4 Genetic Variants in Alport Syndrome: Clinical Characterization of 52 Individuals from 38 Families
- PMID: 40806767
- PMCID: PMC12347723
- DOI: 10.3390/ijms26157639
Lithuanian Study on COL4A3 and COL4A4 Genetic Variants in Alport Syndrome: Clinical Characterization of 52 Individuals from 38 Families
Abstract
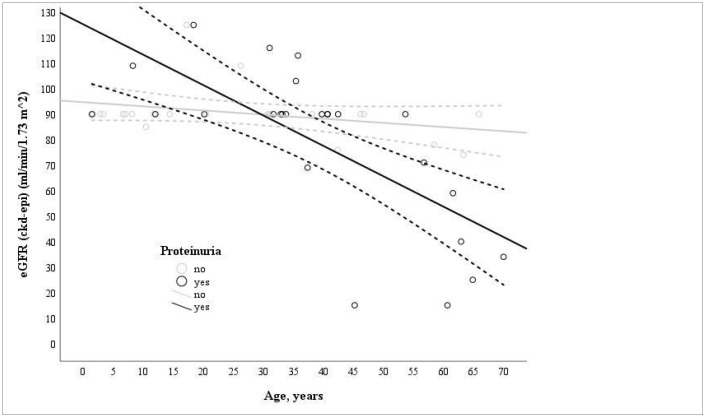
Variants in COL4A3 and COL4A4 cause autosomal dominant and recessive Alport syndrome, yet data on their distribution and clinical expression in different populations remain limited. This study investigated genotype-phenotype correlations and the distribution of COL4A3/COL4A4 variants in a Lithuanian Alport syndrome cohort. A total of 221 individuals from Lithuania were analyzed for COL4A3 and COL4A4 variants using either next-generation sequencing or Sanger sequencing in order to assess variant distribution and associated clinical features. Only individuals with pathogenic, likely pathogenic, or uncertain significance variants were included. Fifty-two individuals (38 index cases) with pathogenic, likely pathogenic, or variants of uncertain significance were identified, as follows: forty-eight were heterozygous, four had autosomal recessive, and four had digenic Alport syndrome. COL4A3 variants were found in 9.5% (21/221) and COL4A4 in 17.6% (39/221). Among the 28 identified variants, 18 were novel. Glycine substitutions (n = 8) were the most frequent and associated with worse kidney outcomes and increased hearing abnormalities. Hematuria was diagnosed significantly earlier than proteinuria (p = 0.05). Most individuals with autosomal dominant Alport syndrome had normal kidney function (eGFR > 90 mL/min/1.73 m2), while those with autosomal recessive Alport syndrome had more severe disease. Kidney failure occurred in 2/4 (50%) autosomal recessive Alport syndrome and 2/48 (4%) autosomal dominant Alport syndrome cases. A significant inverse correlation was found between eGFR and age in proteinuric individuals (r = -0.737; p = 0.013). This study expands knowledge of Alport syndrome in the Lithuanian population and contributes novel variant data to the global Alport syndrome genetic database.
Keywords: Alport syndrome; COL4A3; COL4A4; genotype–phenotype correlation.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Rheault M.N., Kashtan C.E. Pediatric Nephrology. 7th ed. Springer; Berlin/Heidelberg, Germany: 2015. Inherited Glomerular Diseases; pp. 777–803.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
