

Aere perennius: how chromatin fidelity is maintained and lost in disease
- PMID: 40809411
- PMCID: PMC12342192
- DOI: 10.1093/narmme/ugaf026
Aere perennius: how chromatin fidelity is maintained and lost in disease
Abstract
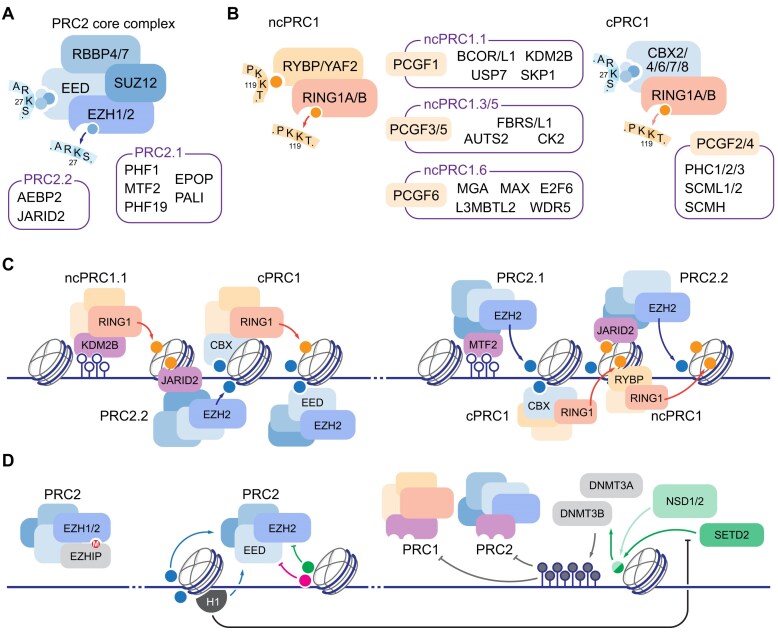
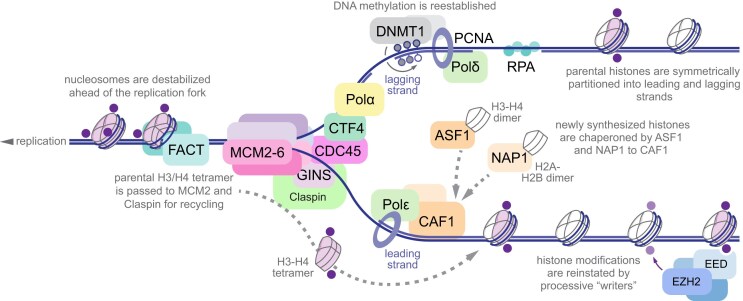
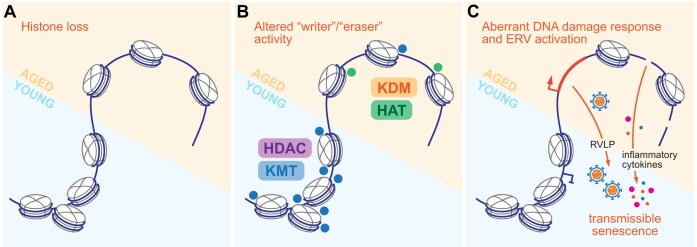
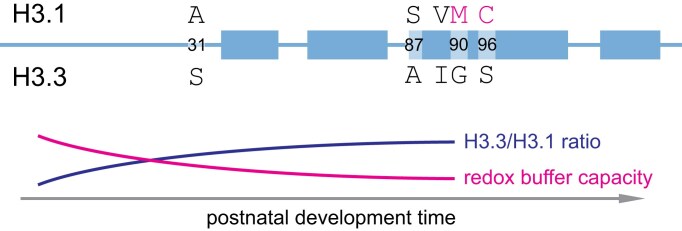
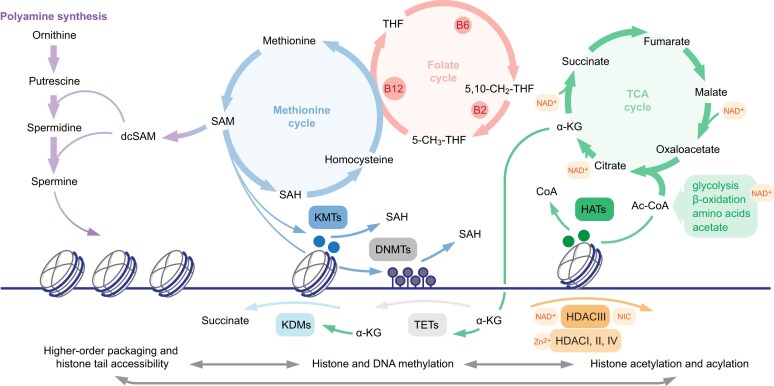
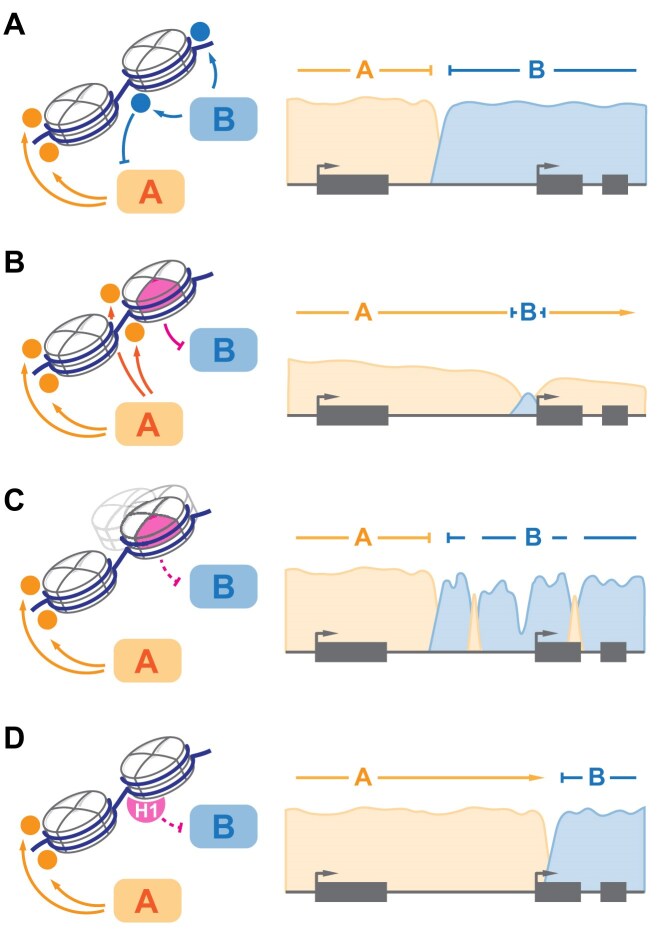
Multicellular organisms arise from a single genome template in the zygote, necessitating the cells of the developing embryo to up- and downregulate specific genes to establish and maintain their identity. This template is maintained, propagated, and interpreted as chromatin, a polymer of nucleic acids and associated structural and regulatory proteins. Recent genome-wide surveys documented a wealth of disease-associated mutations in chromatin factors, indicating their fundamental significance and potential for therapeutic targeting. However, chromatin factors exist in a complex balance, with a single deficiency often leading to pleiotropic downstream effects. Here, we review the mechanisms of chromatin regulation and partitioning, highlighting examples of how these processes are altered in human diseases. We argue that loss of chromatin fidelity, both locally at specific genes and regulatory elements, and globally at the megabase-scale, contributes to many pathological states and may thus represent an intriguing target for corrective interventions.
© The Author(s) 2025. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures
Similar articles
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
How lived experiences of illness trajectories, burdens of treatment, and social inequalities shape service user and caregiver participation in health and social care: a theory-informed qualitative evidence synthesis.Health Soc Care Deliv Res. 2025 Jun;13(24):1-120. doi: 10.3310/HGTQ8159. Health Soc Care Deliv Res. 2025. PMID: 40548558
-
The Black Book of Psychotropic Dosing and Monitoring.Psychopharmacol Bull. 2024 Jul 8;54(3):8-59. Psychopharmacol Bull. 2024. PMID: 38993656 Free PMC article. Review.
-
An Examination of Perceived Stress and Emotion Regulation Challenges as Mediators of Associations Between Camouflaging and Internalizing Symptomatology.Autism Adulthood. 2024 Sep 16;6(3):345-361. doi: 10.1089/aut.2022.0121. eCollection 2024 Sep. Autism Adulthood. 2024. PMID: 39371362
References
Publication types
LinkOut - more resources
Full Text Sources