

Large-scale protein interactome reveals lineage-specific genes driving plant-parasitic nematode adaptive innovations
- PMID: 40815650
- PMCID: PMC12356263
- DOI: 10.1126/sciadv.adt5107
Large-scale protein interactome reveals lineage-specific genes driving plant-parasitic nematode adaptive innovations
Abstract
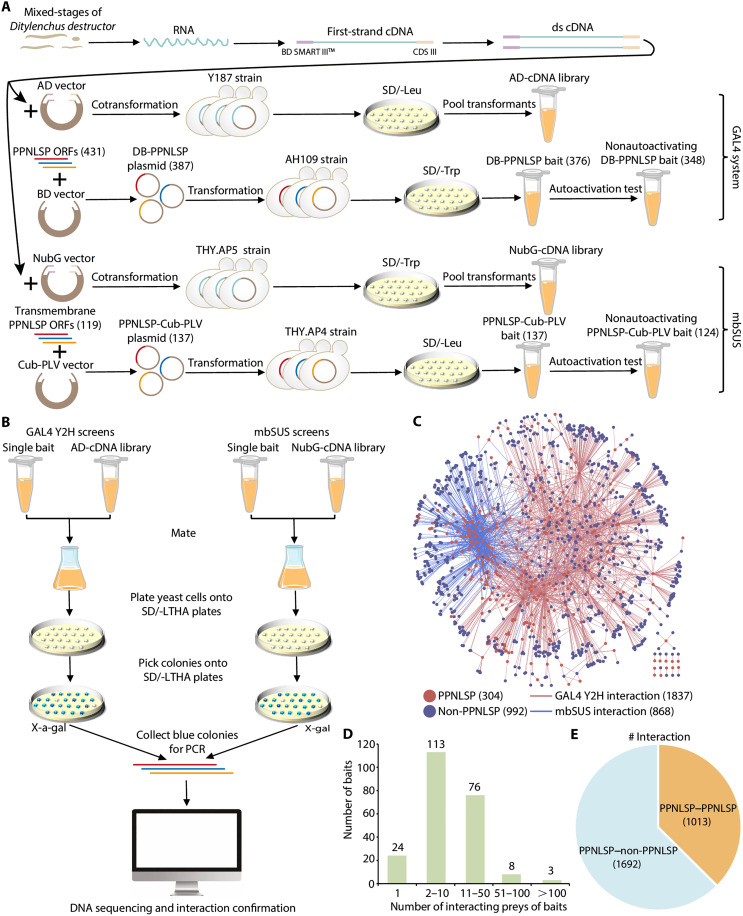
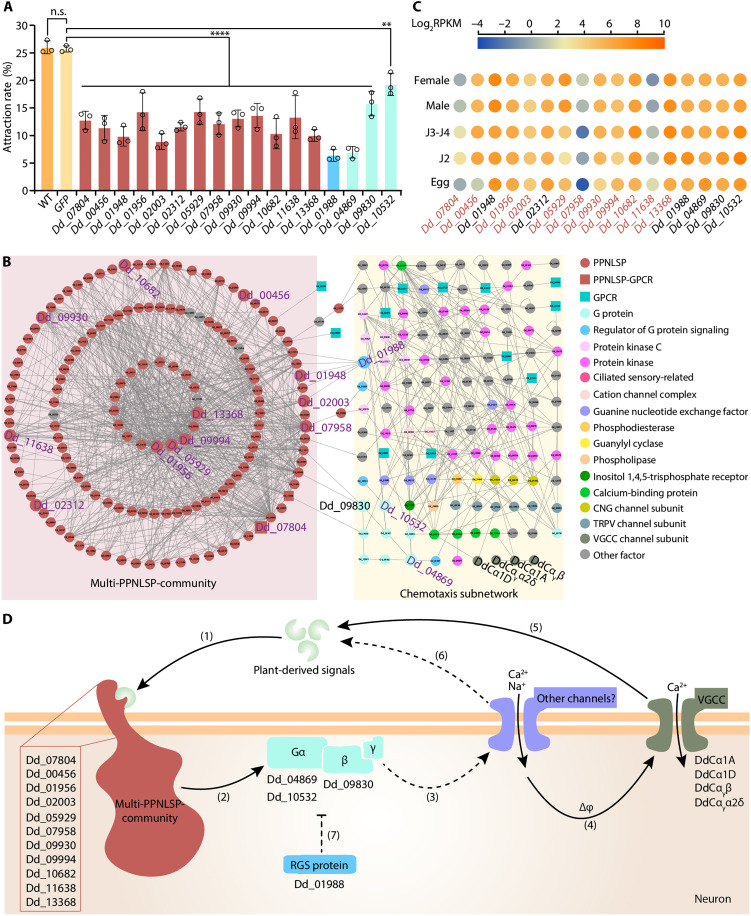
Mounting evidence suggests that lineage-specific genes drive phenotype diversity. Plant-parasitic nematodes (PPNs), among the most destructive plant pathogens, have evolved innovated traits required for plant parasitism, yet the genetic basis remains unclear. Here, we identify PPN lineage-specific genes (PPNLSGs) and analyze the large-scale protein interactome of their encoded proteins (PPNLSPs). By using yeast two-hybrid assays, we identify 2705 protein-protein interactions involving PPNLSPs from stem nematode Ditylenchus destructor, and by using computational methods, we predict conserved interactions of D. destructor proteins at the genome-wide level. Integration of these data allows generating a comprehensive protein interactome map, showing established complexes and PPNLSP modules, and allowing functional annotations for 306 uncharacterized PPNLSPs. Among these interactions, we identify multiple PPNLSPs associated with chemotaxis and infectivity based on these PPNLSP modules and propose a chemotaxis pathway model of host seeking. Our study indicates PPNLSGs as drivers of PPN adaptive innovations and provides a reference resource for future research on PPN biology and control strategies.
Figures





Similar articles
-
Neuronal Basis of Host-Finding and Feeding in Plant-Parasitic Nematodes.Annu Rev Phytopathol. 2025 Sep;63(1):383-402. doi: 10.1146/annurev-phyto-121823-033645. Epub 2025 May 21. Annu Rev Phytopathol. 2025. PMID: 40397805 Review.
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Molecular Insights into Migratory Plant-Parasitic Nematodes.Annu Rev Phytopathol. 2025 Sep;63(1):403-430. doi: 10.1146/annurev-phyto-112624-103056. Annu Rev Phytopathol. 2025. PMID: 40903421 Review.
-
Plant-parasitic nematodes: towards understanding molecular players in stress responses.Ann Bot. 2017 Mar 1;119(5):775-789. doi: 10.1093/aob/mcw260. Ann Bot. 2017. PMID: 28087659 Free PMC article. Review.
-
Tiny Invaders, Big Trouble: Emerging Nematode Threats in the United States.Phytopathology. 2025 Jun;115(6):587-595. doi: 10.1094/PHYTO-09-24-0290-IA. Epub 2025 Jun 6. Phytopathology. 2025. PMID: 39961028 Review.
References
-
- Erwin D. H., Novelty and innovation in the history of life. Curr. Biol. 25, R930–R940 (2015). - PubMed
-
- Wright C. J., Smith C. W. J., Jiggins C. D., Alternative splicing as a source of phenotypic diversity. Nat. Rev. Genet. 23, 697–710 (2022). - PubMed
-
- Kjer-Hansen P., Weatheritt R. J., The function of alternative splicing in the proteome: Rewiring protein interactomes to put old functions into new contexts. Nat. Struct. Mol. Biol. 30, 1844–1856 (2023). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
