

Diversity and composition of sponge-associated microbiomes from Korean sponges revealed by full-length 16S rRNA analysis
- PMID: 40819100
- PMCID: PMC12357920
- DOI: 10.1038/s41598-025-16155-6
Diversity and composition of sponge-associated microbiomes from Korean sponges revealed by full-length 16S rRNA analysis
Abstract
Marine sponges host diverse and specialized microbial communities that serve essential functions in nutrient cycling, ecosystem stability, and biotechnological applications. This study investigates the diversity and composition of sponge-associated microbiomes from eight sponge species collected in Chuksan Harbor, South Korea, using full-length 16S rRNA sequencing and amplicon sequence variant (ASV)-based methods. Our results demonstrate that each sponge species harbors distinct and highly structured microbial communities. Proteobacteria, and especially Alpha- and Gammaproteobacteria, were generally dominant; however, unique dominance patterns, such as the near-exclusive presence of an uncharacterized Gammaproteobacterial lineage in Cliona celata, suggest strong host-symbiont specificity and possible coevolution. Notably, no ASVs were shared between seawater and sponge samples, confirming that sponge hosts select and maintain unique sets of microbial partners. In several Halichondria species, we detected the presence of Entotheonella, a symbiont with high biosynthetic gene cluster diversity that may contribute to host chemical defense and metabolic versatility. Depth-driven differences in microbial community composition were exemplified by Geodia reniformis, whose microbiome was dominated by deep-sea adapted and metabolically versatile lineages such as SAR202, PAUC34f, and Dadabacteriales. This study establishes a new baseline for understanding sponge-microbe partnerships in Korean marine environments. Our integrative, high-resolution approach not only uncovers remarkable taxonomic and functional diversity, but also provides a valuable genetic resource for future marine natural-product discovery and advances ecological restoration efforts.
Keywords: Amplicon sequencing; Full-length 16S rRNA; Korean sponges; Microbial community composition; Sponge microbiome; Symbiotic microorganisms.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures

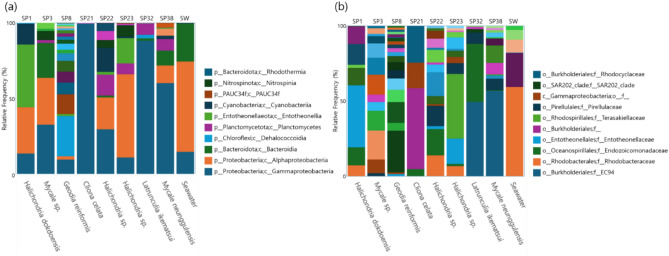


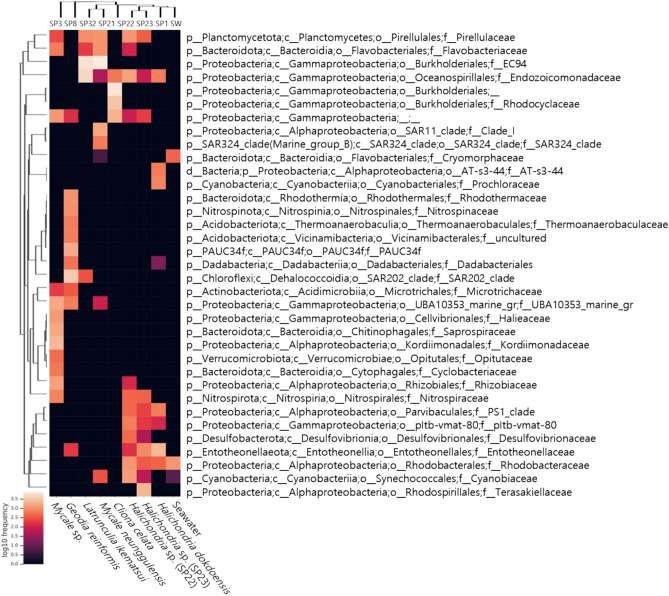
Similar articles
-
Sponges from Lembeh Strait: antimicrobial activity and prokaryotic diversity.J Appl Microbiol. 2025 Jun 2;136(6):lxaf129. doi: 10.1093/jambio/lxaf129. J Appl Microbiol. 2025. PMID: 40408290
-
Evaluation of V3-V4 and FL-16S rRNA amplicon sequencing approach for microbiota community analysis of tracheostomy aspirates.mSphere. 2025 Aug 26;10(8):e0038825. doi: 10.1128/msphere.00388-25. Epub 2025 Jul 22. mSphere. 2025. PMID: 40693760 Free PMC article.
-
The microbiome of the sponge Aplysina caissara in two sites with different levels of anthropogenic impact.FEMS Microbiol Lett. 2023 Jan 17;370:fnad064. doi: 10.1093/femsle/fnad064. FEMS Microbiol Lett. 2023. PMID: 37401172
-
Bacterial diversity in the freshwater sponges of Sundarban and their potential role in biomonitoring toxic element pollution.Microbiol Spectr. 2025 Aug 27:e0214925. doi: 10.1128/spectrum.02149-25. Online ahead of print. Microbiol Spectr. 2025. PMID: 40862605
-
High-throughput identification and quantification of bacterial cells in the microbiota based on 16S rRNA sequencing with single-base accuracy using BarBIQ.Nat Protoc. 2024 Jan;19(1):207-239. doi: 10.1038/s41596-023-00906-8. Epub 2023 Nov 27. Nat Protoc. 2024. PMID: 38012397 Review.
References
-
- Hentschel, U., Usher, K. M. & Taylor, M. W. Marine sponges as microbial fermenters. Fems. Microbiol. Ecol.55, 167–177 (2006). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
