

Mitochondrial Quality Control in Health and Disease
- PMID: 40821693
- PMCID: PMC12356995
- DOI: 10.1002/mco2.70319
Mitochondrial Quality Control in Health and Disease
Abstract
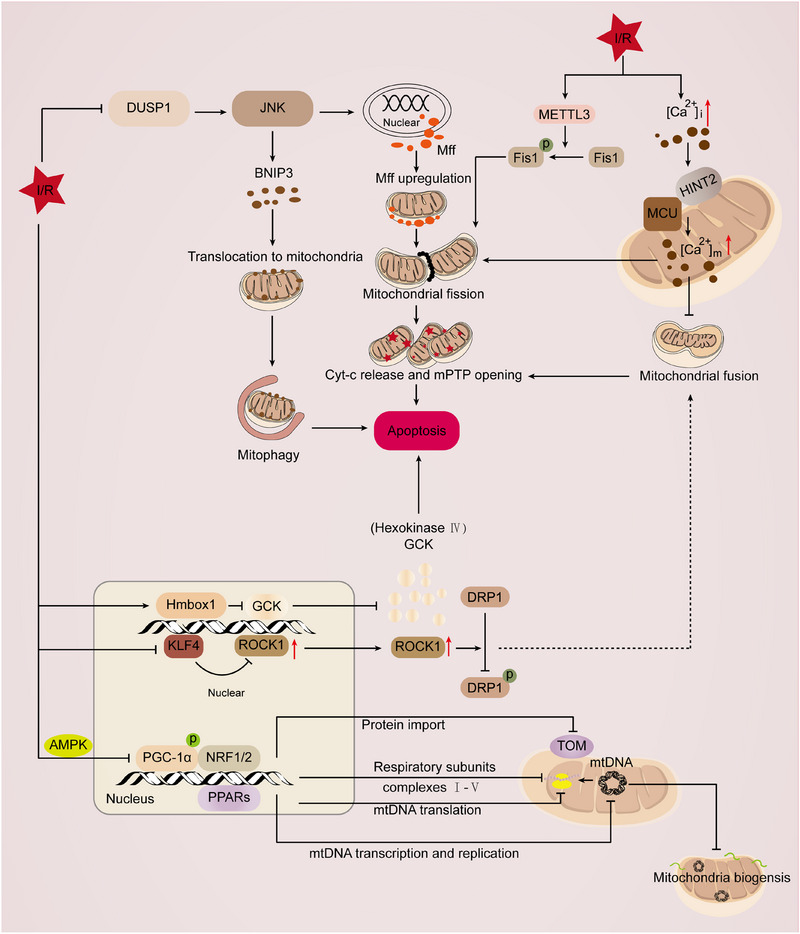
Mitochondria are central regulators of cellular energy metabolism, and their functional integrity is essential for maintaining cellular homeostasis. Mitochondrial quality control (MQC) encompasses a coordinated network of mitochondrial biogenesis, dynamics (fusion and fission), and selective autophagy (mitophagy), which together sustain mitochondrial structure and function. Under physiological conditions, MQC ensures the removal of dysfunctional mitochondria, restricts excessive reactive oxygen species production, and modulates apoptosis, thereby supporting the high energy demands of organs such as the heart and brain. Disruption of MQC contributes to the onset and progression of various diseases, including neurodegenerative disorders, cardiovascular pathologies, and metabolic syndromes, largely through accumulation of damaged mitochondria and impaired metabolic signaling. While the core components of MQC have been characterized, the mechanistic interplay among its modules and their disease-specific alterations remain incompletely defined. This review provides an integrated overview of the molecular pathways governing mitochondrial biogenesis, dynamics, and mitophagy, with a focus on their cross-talk in maintaining mitochondrial homeostasis. We further discuss how MQC dysfunction contributes to disease pathogenesis and examine emerging therapeutic approaches aimed at restoring mitochondrial quality. Understanding the regulatory logic of MQC not only elucidates fundamental principles of cellular stress adaptation but also informs novel strategies for disease intervention.
Keywords: disease intervention; mitochondria; mitochondrial quality control; therapeutic strategies.
© 2025 The Author(s). MedComm published by Sichuan International Medical Exchange & Promotion Association (SCIMEA) and John Wiley & Sons Australia, Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

Similar articles
-
Mitochondrial quality control in hematopoietic stem cells: mechanisms, implications, and therapeutic opportunities.Stem Cell Res Ther. 2025 Apr 15;16(1):180. doi: 10.1186/s13287-025-04304-7. Stem Cell Res Ther. 2025. PMID: 40234908 Free PMC article. Review.
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Mitochondrial dynamics dysfunction and neurodevelopmental disorders: From pathological mechanisms to clinical translation.Neural Regen Res. 2025 Jun 19. doi: 10.4103/NRR.NRR-D-24-01422. Online ahead of print. Neural Regen Res. 2025. PMID: 40537021
-
Role of mitochondria in physiological activities, diseases, and therapy.Mol Biomed. 2025 Jun 19;6(1):42. doi: 10.1186/s43556-025-00284-5. Mol Biomed. 2025. PMID: 40536597 Free PMC article. Review.
-
Role of AMBRA1 in mitophagy regulation: emerging evidence in aging-related diseases.Autophagy. 2024 Dec;20(12):2602-2615. doi: 10.1080/15548627.2024.2389474. Epub 2024 Sep 2. Autophagy. 2024. PMID: 39113560 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources