

Deciphering the proteome of Escherichia coli K-12: Integrating transcriptomics and machine learning to annotate hypothetical proteins
- PMID: 40821719
- PMCID: PMC12356324
- DOI: 10.1016/j.csbj.2025.07.036
Deciphering the proteome of Escherichia coli K-12: Integrating transcriptomics and machine learning to annotate hypothetical proteins
Abstract
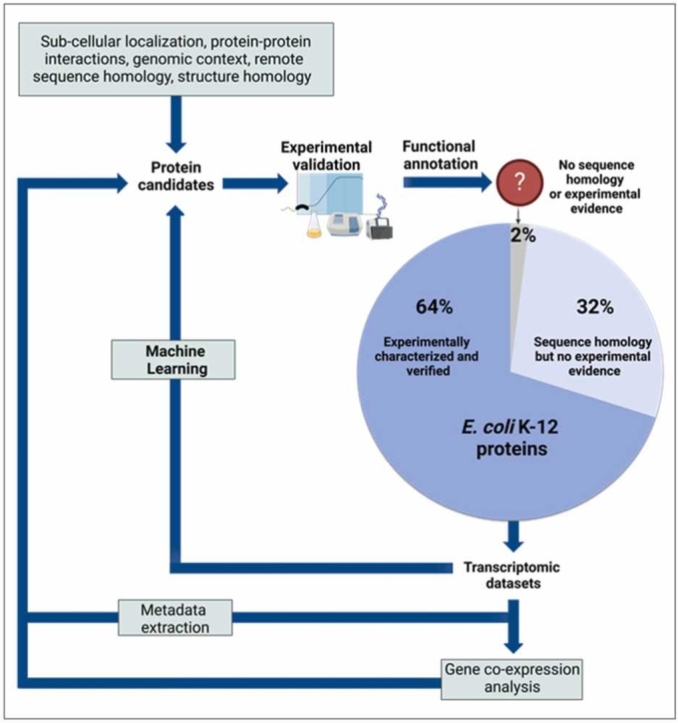
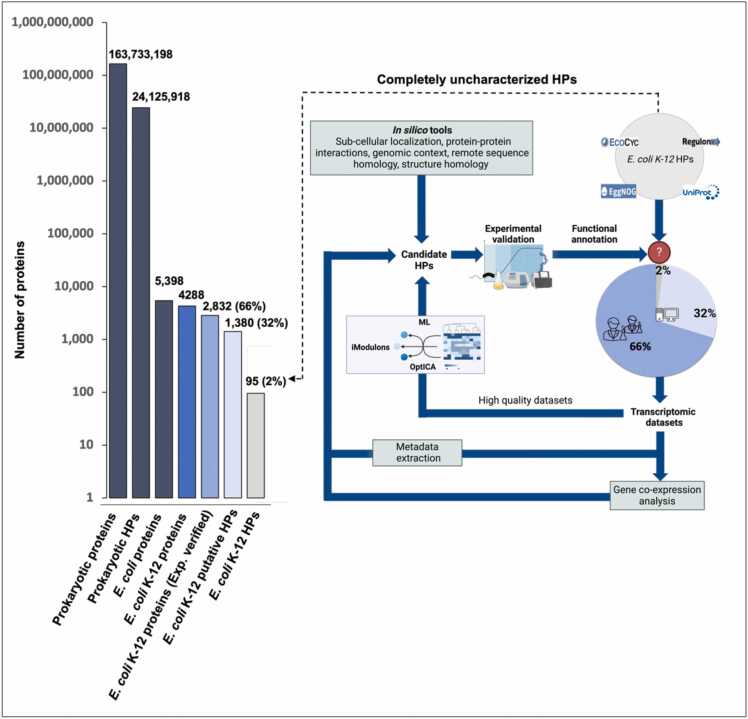
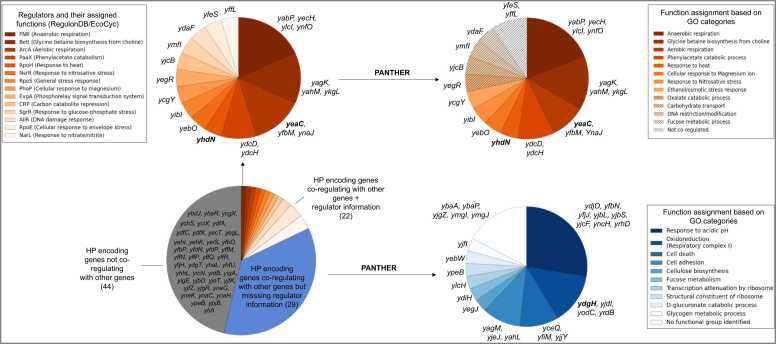
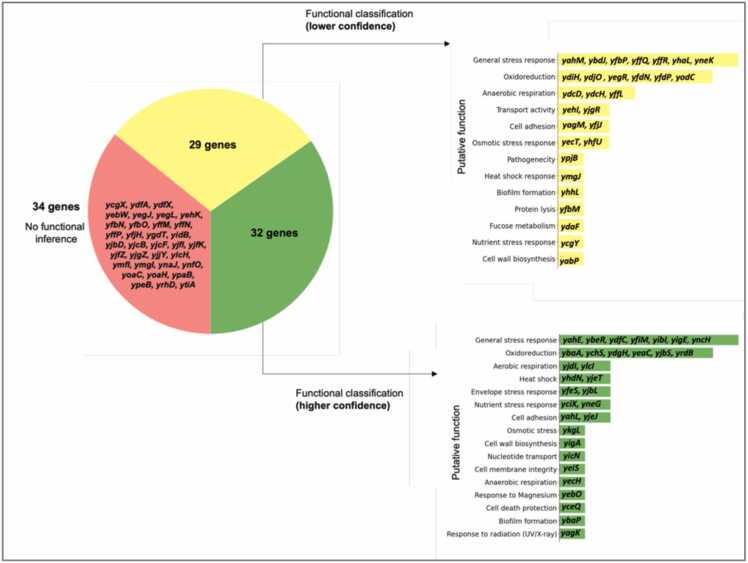
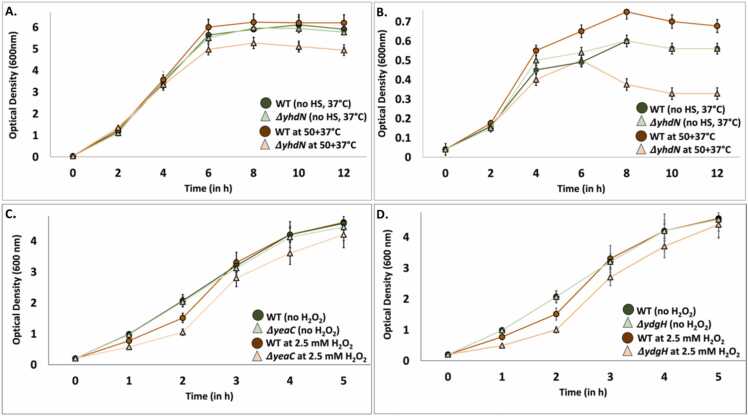
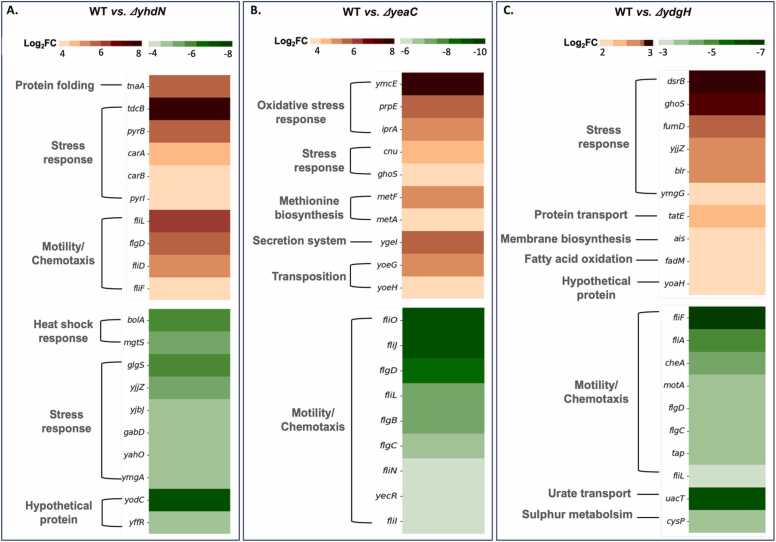
Omics technologies have led to the discovery of a vast number of proteins that are expressed but have no functional annotation - so called hypothetical proteins (HPs). Even in the best-studied model organism Escherichia coli K-12, over 2 % of the proteome remains uncharacterized. This knowledge gap becomes even worse when looking at microbial dark matter. However, knowing the functions of proteins is crucial for elucidating cellular and metabolic processes and harnessing biotechnological potentials. Here, we employed machine learning to decipher the transcriptional regulatory network of E. coli K-12, as well as other in silico tools to assign functions to uncharacterized HPs. We further provide experimental validation of in silico predicted functions for three HP-encoding genes (yhdN, yeaC and ydgH) as proof of concept, by analyzing growth patterns of deletion mutants compared to the wild type, as well as their transcriptional responses to specific conditions. This study demonstrates that the use of Big Omics Data in combination with Artificial Intelligence and experimental controls is a powerful approach to illuminate functional dark matter.
Keywords: Artificial intelligence; Big omics data; Functional annotation of proteins; Functional dark matter; Independent Component Analysis (ICA).
© 2025 The Authors.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Behavioral interventions to reduce risk for sexual transmission of HIV among men who have sex with men.Cochrane Database Syst Rev. 2008 Jul 16;(3):CD001230. doi: 10.1002/14651858.CD001230.pub2. Cochrane Database Syst Rev. 2008. PMID: 18646068
-
EORTC guidelines for the use of erythropoietic proteins in anaemic patients with cancer: 2006 update.Eur J Cancer. 2007 Jan;43(2):258-70. doi: 10.1016/j.ejca.2006.10.014. Epub 2006 Dec 19. Eur J Cancer. 2007. PMID: 17182241
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous