

Intrinsic disorder in CYP1B1 and its implications in primary congenital glaucoma pathogenesis
- PMID: 40821877
- PMCID: PMC12352203
- DOI: 10.1007/s42485-025-00186-8
Intrinsic disorder in CYP1B1 and its implications in primary congenital glaucoma pathogenesis
Abstract
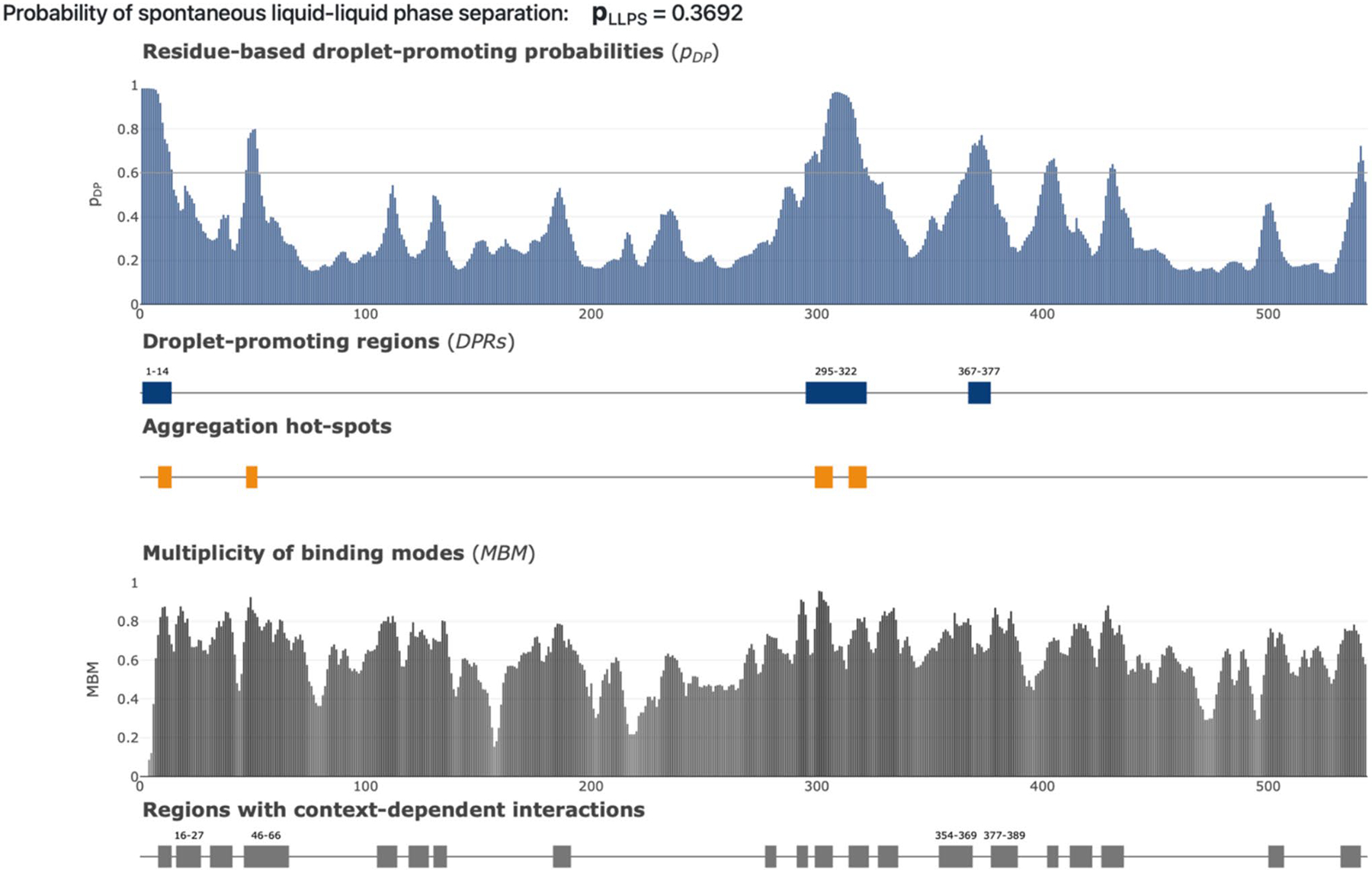
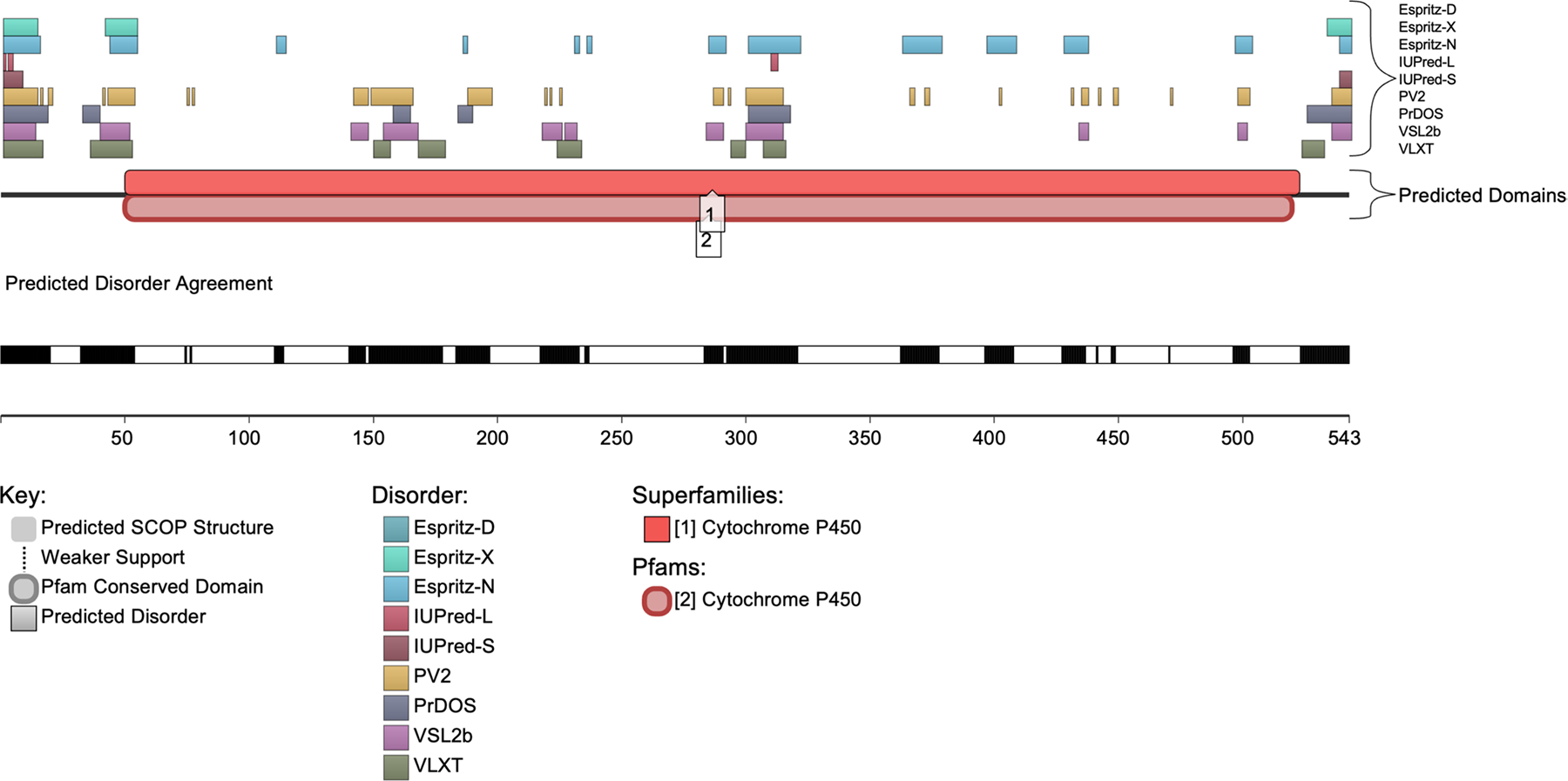
Cytochrome P450 1B1 (CYP1B1) plays a critical role in the pathogenesis of primary congenital glaucoma (PCG), a severe eye disorder that can lead to pediatric blindness if untreated. Increasing evidence suggests that intrinsically disordered proteins and regions (IDPs/IDPRs), which lack a stable three-dimensional structure, are significant in disease pathology due to their flexible nature, impacting protein interactions and function. This study explores the intrinsic disorder within CYP1B1 and its implications in the molecular mechanisms underlying PCG. We employed a comprehensive bioinformatics approach to assess the structural and functional properties of CYP1B1 using tools such as AlphaMissense, a tool crafted to evaluate the functional impact of missense mutations in proteins. Our structural analysis qualitatively demonstrated that CYP1B1 contains intrinsically disordered protein regions (i.e., spaghetti-like entities) that are structureless and flexible. Correlation analysis showed that disorder decreases exponentially relative to AlphaMissense predicted pathogenicity, with an exponential decay fit (R 2 = 0.62), suggesting that highly disordered regions tend to harbor benign mutations. This study identifies critical intrinsically disordered regions within CYP1B1 and elucidates its complex interaction network, highlighting the potential role of these regions in PCG pathogenesis. The observed correlation between intrinsic disorder and reduced pathogenicity of mutations suggests that IDPRs may buffer against deleterious effects, providing a possible explanation for the variability in clinical outcomes associated with CYP1B1 mutations. These insights enhance our understanding of the molecular basis of PCG and offer potential targets for novel therapeutic interventions to combat this blinding childhood disorder.
Keywords: AlphaFold; AlphaMissense; D2P2; FuzDrop; RIDAO; STRING.
Conflict of interest statement
Conflict of interest The authors declare no competing interests.
Figures





Similar articles
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
The effects of retinal disease on intrinsic protein disorder and liquid-liquid‑phase separation.J Proteins Proteom. 2025 Jun 19:10.1007/s42485-025-00188-6. doi: 10.1007/s42485-025-00188-6. Online ahead of print. J Proteins Proteom. 2025. PMID: 40799657 Free PMC article.
-
Bioinformatics-Based Comparative Analysis of the Human Retina Proteome.Proteomics Clin Appl. 2025 Jul;19(4):e70012. doi: 10.1002/prca.70012. Epub 2025 Jun 7. Proteomics Clin Appl. 2025. PMID: 40481689 Free PMC article.
-
Management of urinary stones by experts in stone disease (ESD 2025).Arch Ital Urol Androl. 2025 Jun 30;97(2):14085. doi: 10.4081/aiua.2025.14085. Epub 2025 Jun 30. Arch Ital Urol Androl. 2025. PMID: 40583613 Review.
-
Gender differences in the context of interventions for improving health literacy in migrants: a qualitative evidence synthesis.Cochrane Database Syst Rev. 2024 Dec 12;12(12):CD013302. doi: 10.1002/14651858.CD013302.pub2. Cochrane Database Syst Rev. 2024. PMID: 39665382
References
Grants and funding
LinkOut - more resources
Full Text Sources