

Internal ribosome entry sites enhance translation in trans in antisense non-coding SINEUP and circular RNAs
- PMID: 40823812
- PMCID: PMC12359042
- DOI: 10.1093/nar/gkaf788
Internal ribosome entry sites enhance translation in trans in antisense non-coding SINEUP and circular RNAs
Abstract
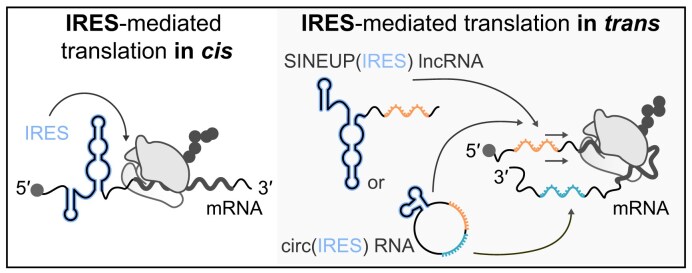
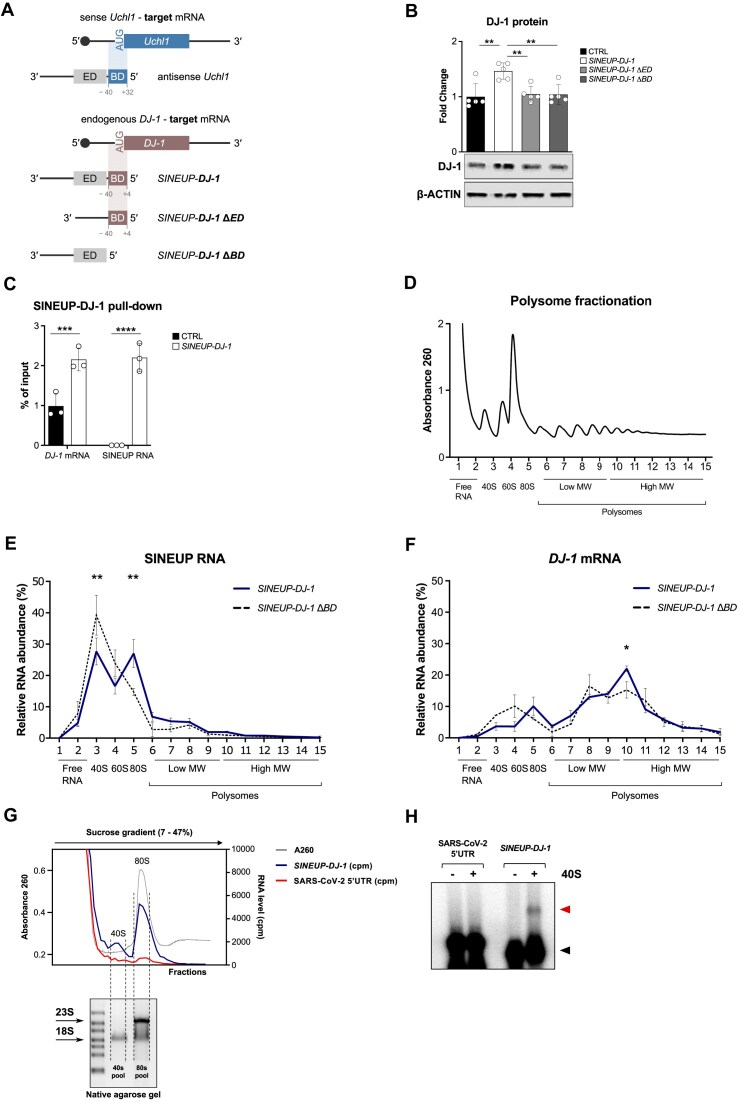
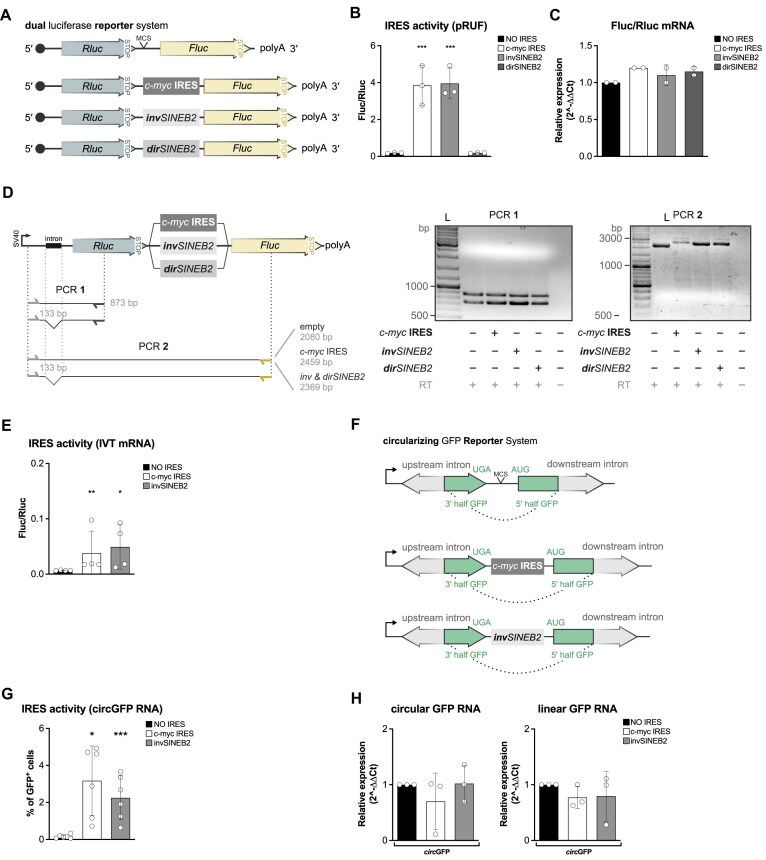
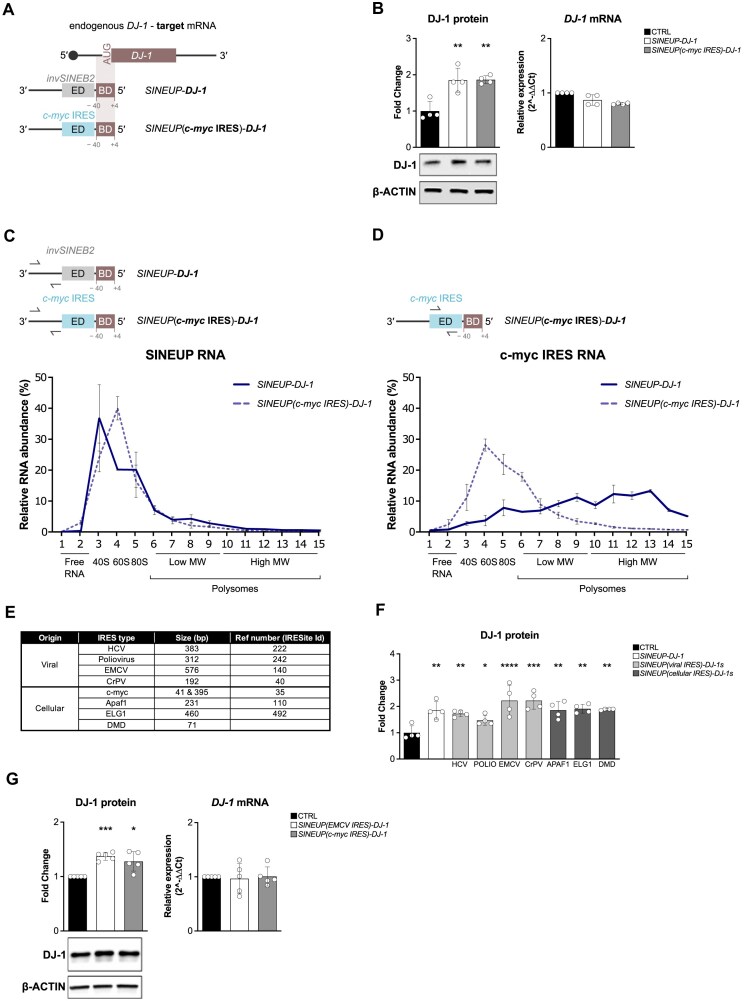
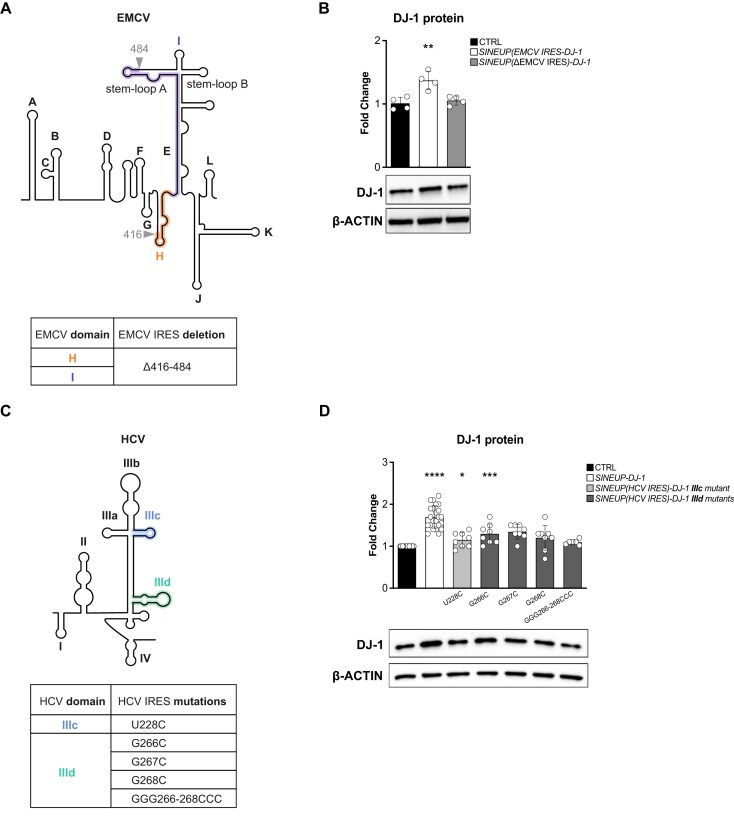
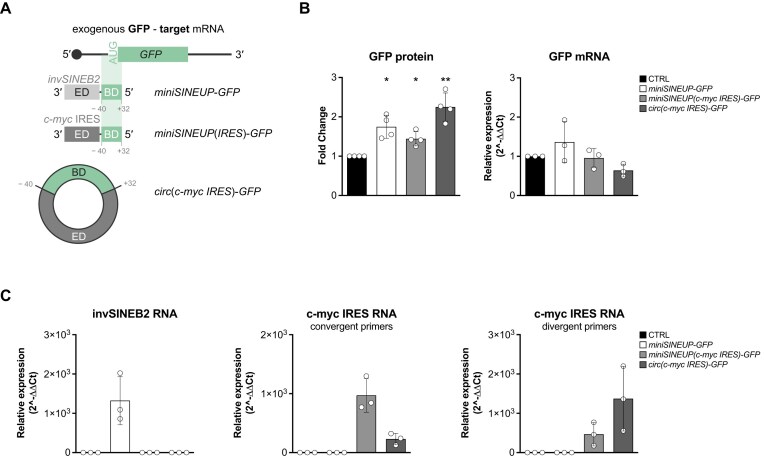
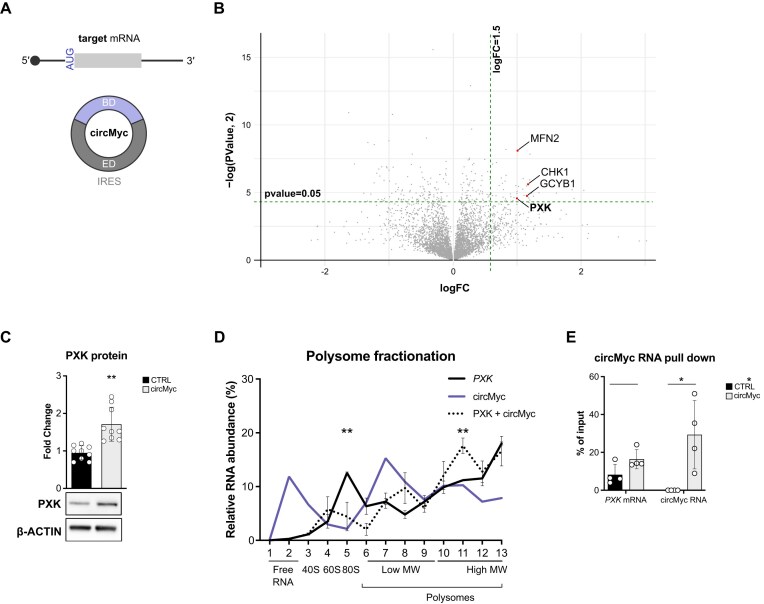
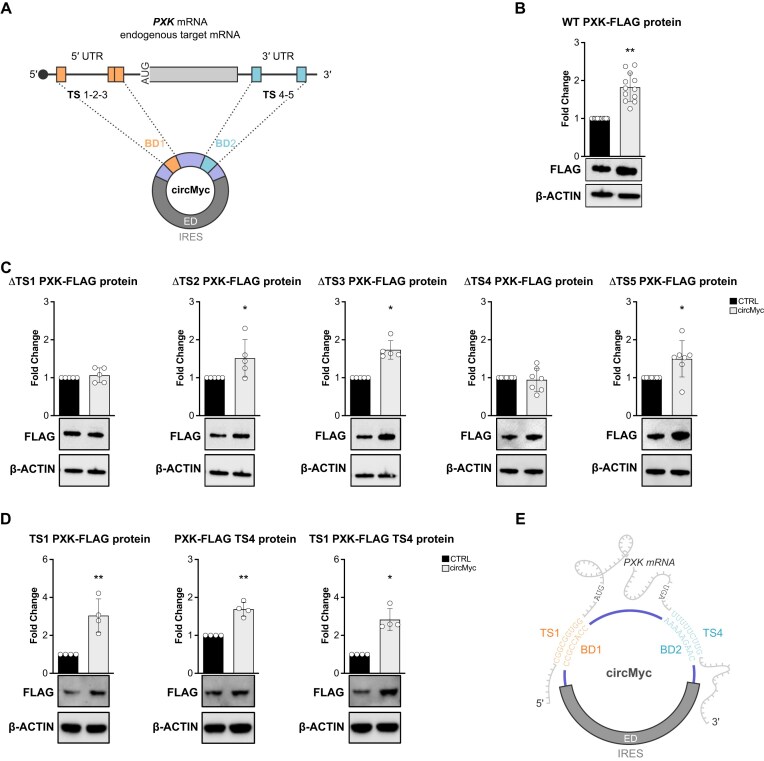
Sequences in the 5'-untranslated regions of cellular and viral mRNAs can function as internal ribosome entry sites (IRESs), driving cis-acting translation of the downstream protein-coding open reading frame. Here we demonstrate that RNA sequences with either newly identified or well-characterized IRES activity can also induce trans-acting translation of an independent mRNA species through an antisense sequence. SINEUPs are antisense long non-coding RNAs that enhance the translation of overlapping sense mRNAs in trans by employing two critical domains: the invSINEB2 sequence, which up-regulates translation (effector domain), and an antisense region providing target specificity (binding domain). First, we show that the invSINEB2 from the natural SINEUP AS Uchl1 RNA acts as an IRES when functioning in cis. Next, we establish that known viral and cellular sequences with IRES activity can operate in trans as an effector domain in synthetic SINEUPs. To identify natural IRES-containing non-coding RNAs with transactivity, we found that the non-coding hsa_circ_0 085 533 (circMyc), transcribed from the c-myc locus, enhances protein expression of PX Domain Containing Serine/Threonine Kinase Like (PXK) by promoting mRNA association with polysomes through antisense sequences. These results suggest that SINEUPs and some circular RNAs are trans-acting IRESs, expanding the repertoire of molecular mechanisms to regulate translation.
© The Author(s) 2025. Published by Oxford University Press.
Conflict of interest statement
S.G., S.Z., P.C., and C.S. declare competing financial interests as co-founders of Harness Therapeutics, UK. S.G. and S.D. are co-inventors of a patent on the use of circular SINEUPs for therapy. S.G. and S.Z. are co-inventors of a patent covering the use of IRESs as an effector domain in SINEUPs.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
