

Macrophages and macrophage extracellular vesicles confer cancer ferroptosis resistance via PRDX6-mediated mitophagy inhibition
- PMID: 40825268
- PMCID: PMC12391288
- DOI: 10.1016/j.redox.2025.103826
Macrophages and macrophage extracellular vesicles confer cancer ferroptosis resistance via PRDX6-mediated mitophagy inhibition
Abstract
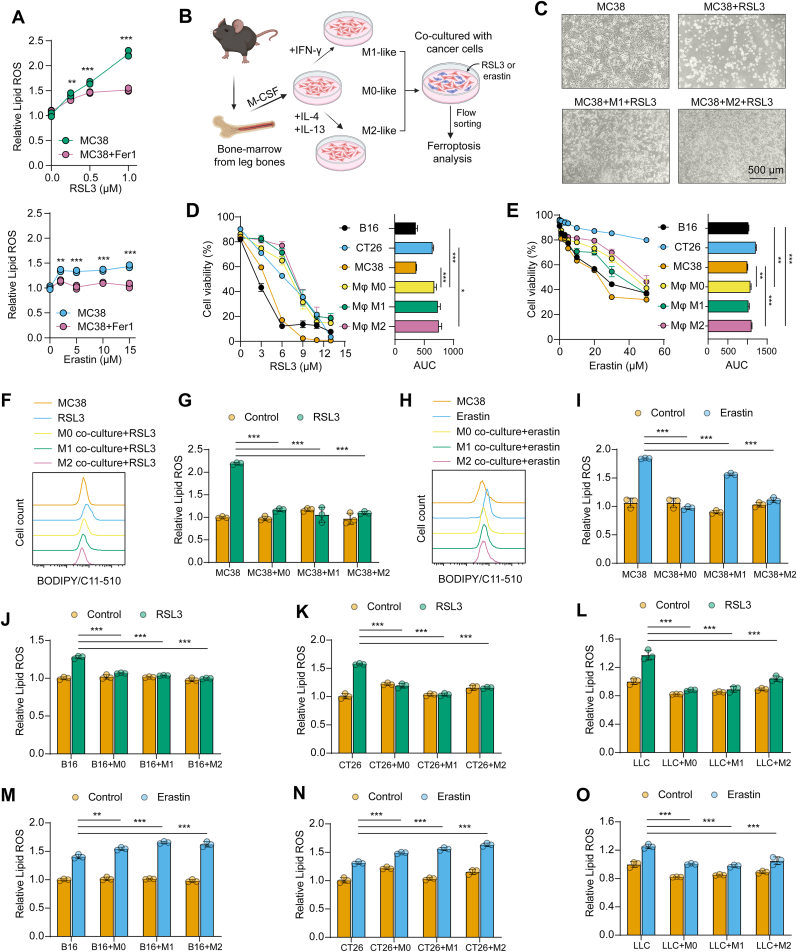
Ferroptosis has emerged as a promising therapeutic target in cancer therapy, with the tumor microenvironment (TME) playing a pivotal role in regulating ferroptosis. Although macrophages contribute to ferroptosis regulation within TME, the underlying mechanisms remain unclear. In this study, we demonstrate that macrophages consistently attenuate GPX4 inhibitor-induced lipid peroxidation and cell death in various tumor cell lines, whereas their resistance to cysteine transport inhibitor-triggered ferroptosis varies across cell types. This tumor protection from ferroptosis is mediated through macrophage-tumor cell contact and the delivery of macrophage-derived extracellular vesicles (Mφ-EV). Transcriptomic and proteomic analyses revealed that macrophages and Mφ-EV enhance glutathione metabolism in tumor cells. Notably, Mφ-EV are uniquely enriched with the glutathione metabolism-related protein PRDX6. Mechanistically, the glutathione peroxidase activity of PRDX6 elevates intracellular reduced glutathione, suppresses lipid peroxidation, and thereby mitigates ferroptosis. Furthermore, macrophage-derived PRDX6 reduces mitochondrial superoxide accumulation, alleviates ferroptosis-induced mitophagy, and enhances tumor cell viability, ultimately promoting tumor growth. Together, our findings provide a novel mechanism of ferroptosis resistance in TME, wherein macrophages confer tumor cell resilience by bypassing GPX4 inhibition.
Keywords: Extracellular vesicles; Ferroptosis; Macrophages; Mitophagy; Peroxiredoxin 6.
Copyright © 2025. Published by Elsevier B.V.
Conflict of interest statement
Declaration of competing interest Authors declare no conflict of interest.
Figures







Similar articles
-
M1 Macrophage-Derived TNF-α Promotes Pancreatic Cancer Ferroptosis Via p38 MAPK-ACSL4 Pathway.Curr Mol Med. 2025 Jul 10. doi: 10.2174/0115665240374551250630075409. Online ahead of print. Curr Mol Med. 2025. PMID: 40653839
-
Deficiency of PLA2G6 Induces Ferroptosis in Dopaminergic Neurons and Contributes to Parkinson's Disease Pathogenesis via Disruption of PRDX6/FTH1/GPX4 Axis.Mol Neurobiol. 2025 Jul 28. doi: 10.1007/s12035-025-05257-x. Online ahead of print. Mol Neurobiol. 2025. PMID: 40721924
-
Paclitaxel Attenuates Atherosclerosis by Suppressing Macrophage Ferroptosis and Improving Lipid Metabolism via the Sirt1/Nrf2/GPX4 Pathway.FASEB J. 2025 Aug 15;39(15):e70917. doi: 10.1096/fj.202501047RR. FASEB J. 2025. PMID: 40779351
-
Methyltransferase ZC3H13 regulates ferroptosis of alveolar macrophages in sepsis-associated acute lung injury via PRDX6/p53/SLC7A11 axis.Funct Integr Genomics. 2025 Jul 12;25(1):156. doi: 10.1007/s10142-025-01659-1. Funct Integr Genomics. 2025. PMID: 40646387
-
Targeting Ferroptosis in Tumors: Novel Marine-Derived Compounds as Regulators of Lipid Peroxidation and GPX4 Signaling.Mar Drugs. 2025 Jun 19;23(6):258. doi: 10.3390/md23060258. Mar Drugs. 2025. PMID: 40559667 Free PMC article. Review.
References
-
- Liu K., et al. Ferroptosis: the entanglement between traditional drugs and nanodrugs in tumor therapy. Adv. Healthcare Mater. 2023;12(12) - PubMed
-
- Roh J.-L., et al. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016;381(1):96–103. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
