

LIPA, a risk locus for coronary artery disease: decoding the variant-to-function relationship
- PMID: 40827730
- PMCID: PMC12461605
- DOI: 10.1093/eurheartj/ehaf581
LIPA, a risk locus for coronary artery disease: decoding the variant-to-function relationship
Abstract
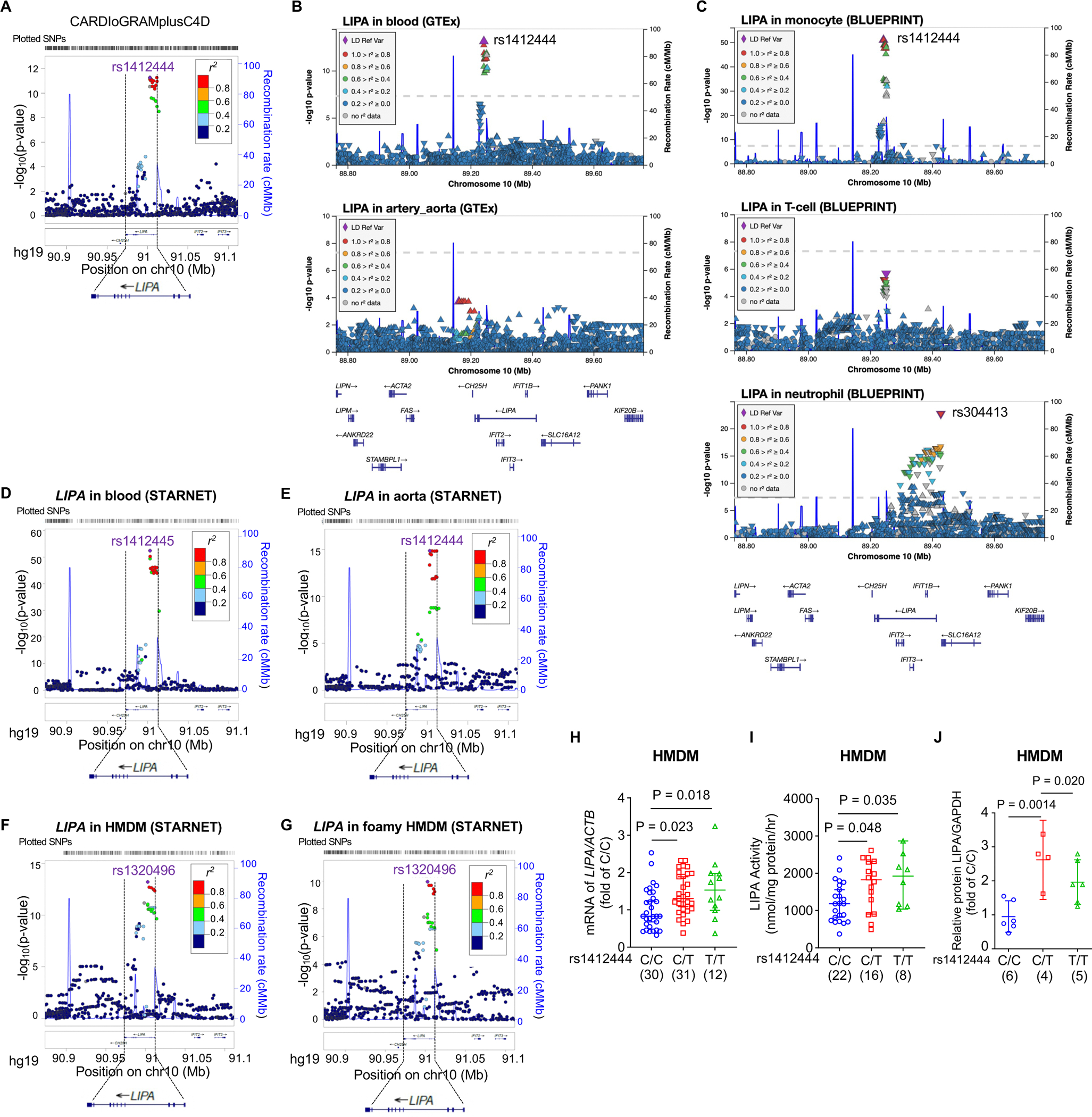
Background and aims: Translating human genomic discoveries into mechanistic insights requires linking genetic variations to candidate genes and their causal functional phenotypes. Genome-wide association studies have consistently identified LIPA (lipase A, lysosomal acid type) as a risk locus for coronary artery disease, with previous analyses prioritising LIPA as a likely causal gene. However, functional studies elucidating causal variants, regulatory mechanisms, target cell types, and their causal impact on atherosclerosis have been lacking. This study aims to address this gap by establishing the variant-to-function relationship at the LIPA locus.
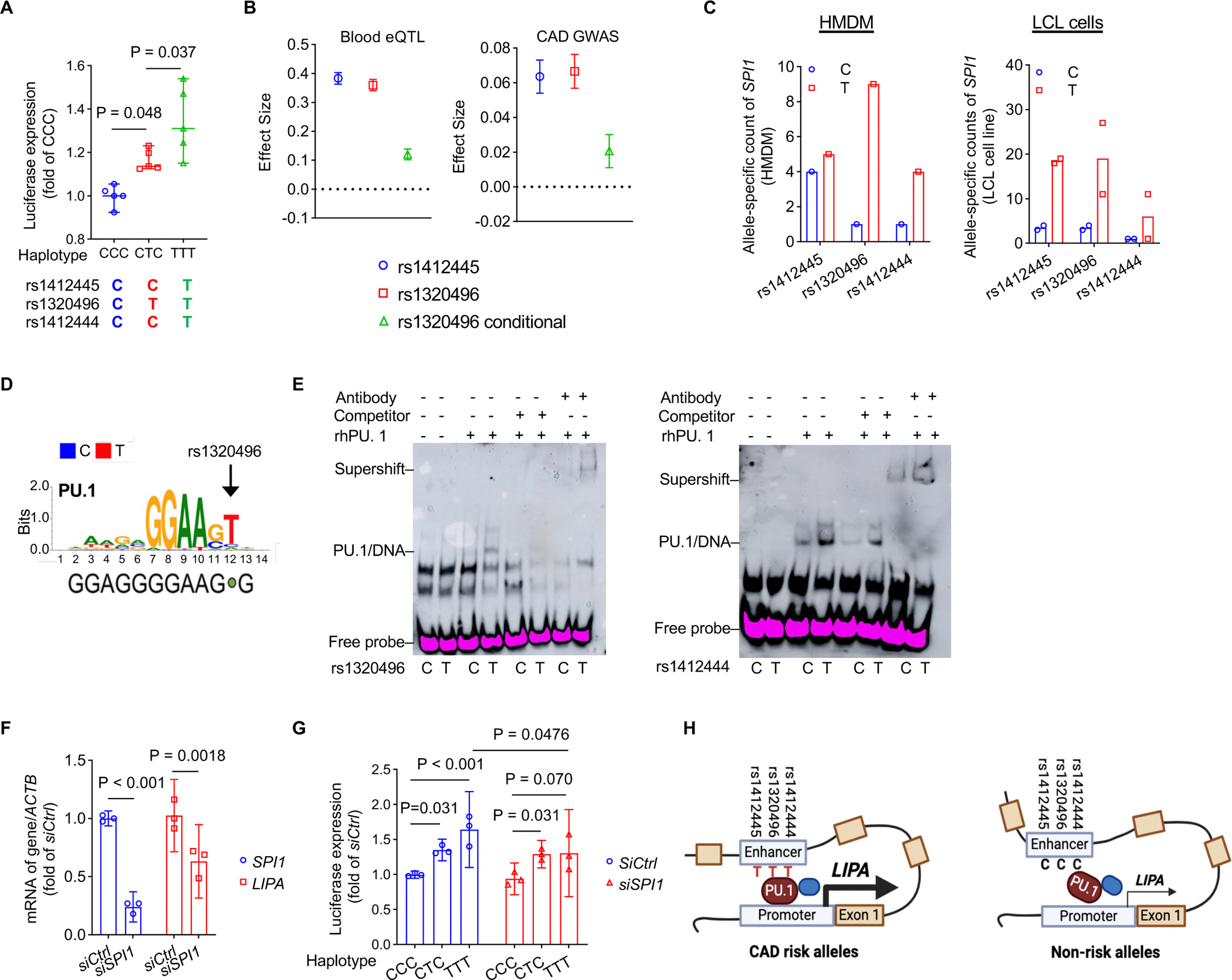
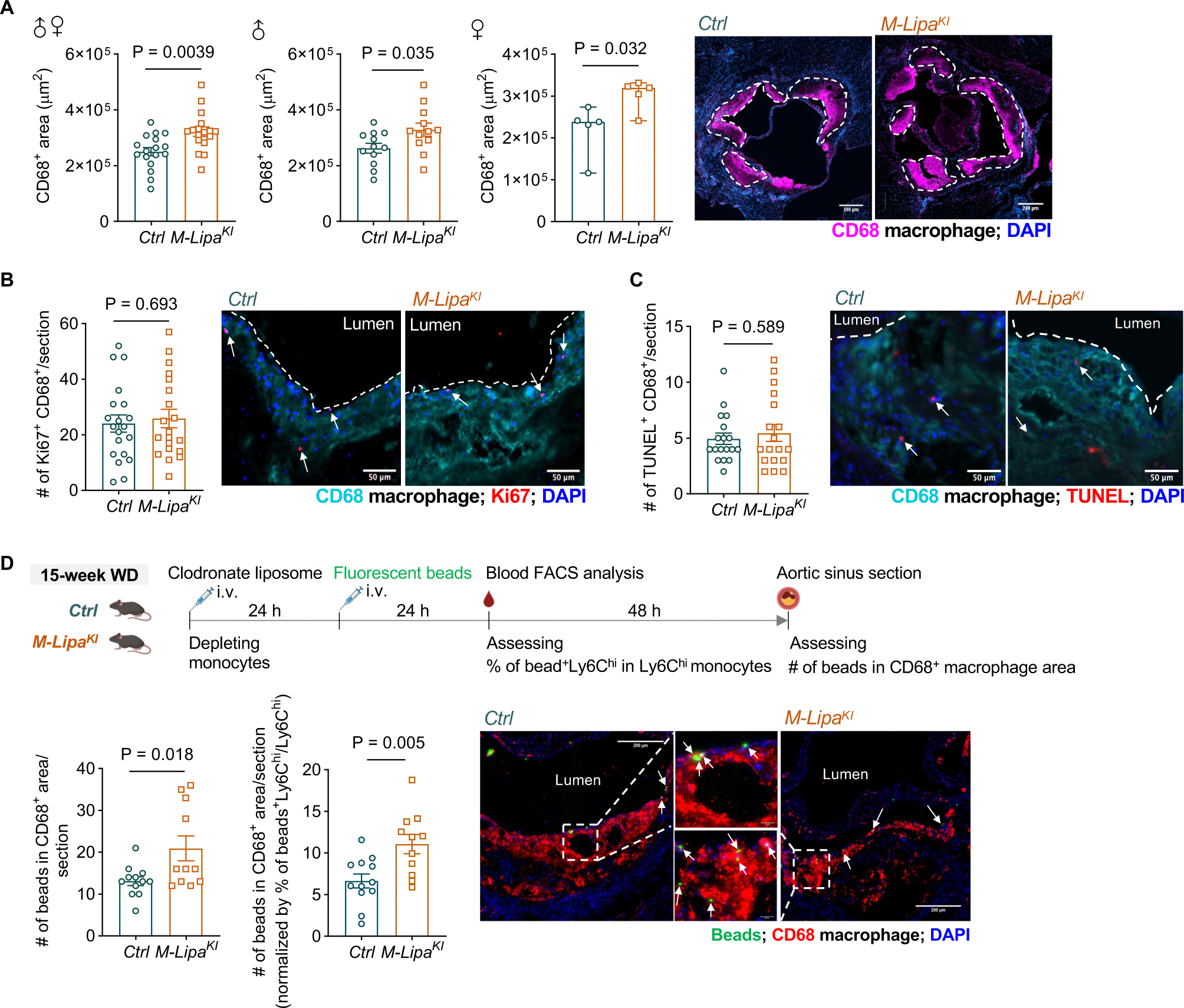
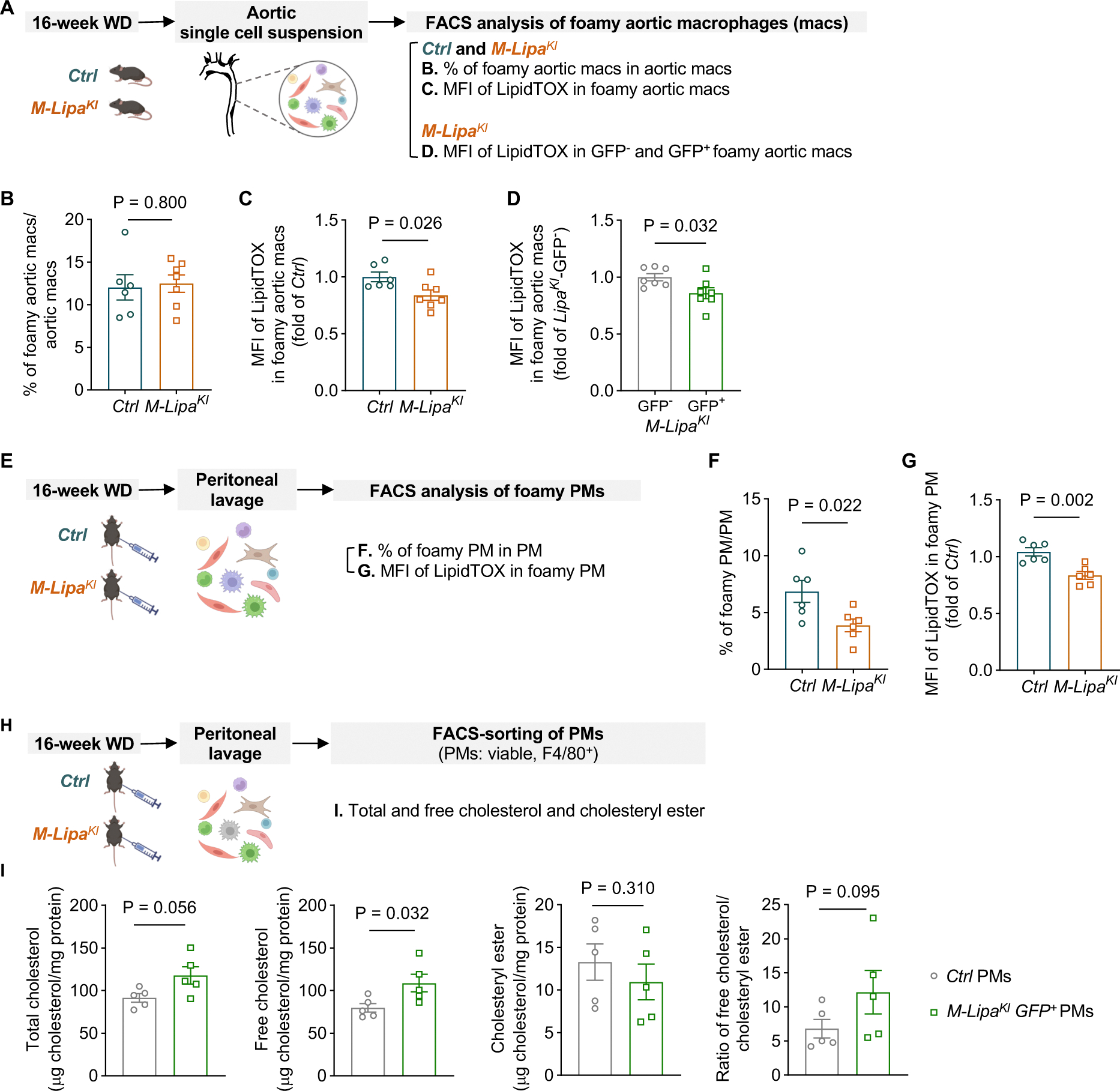
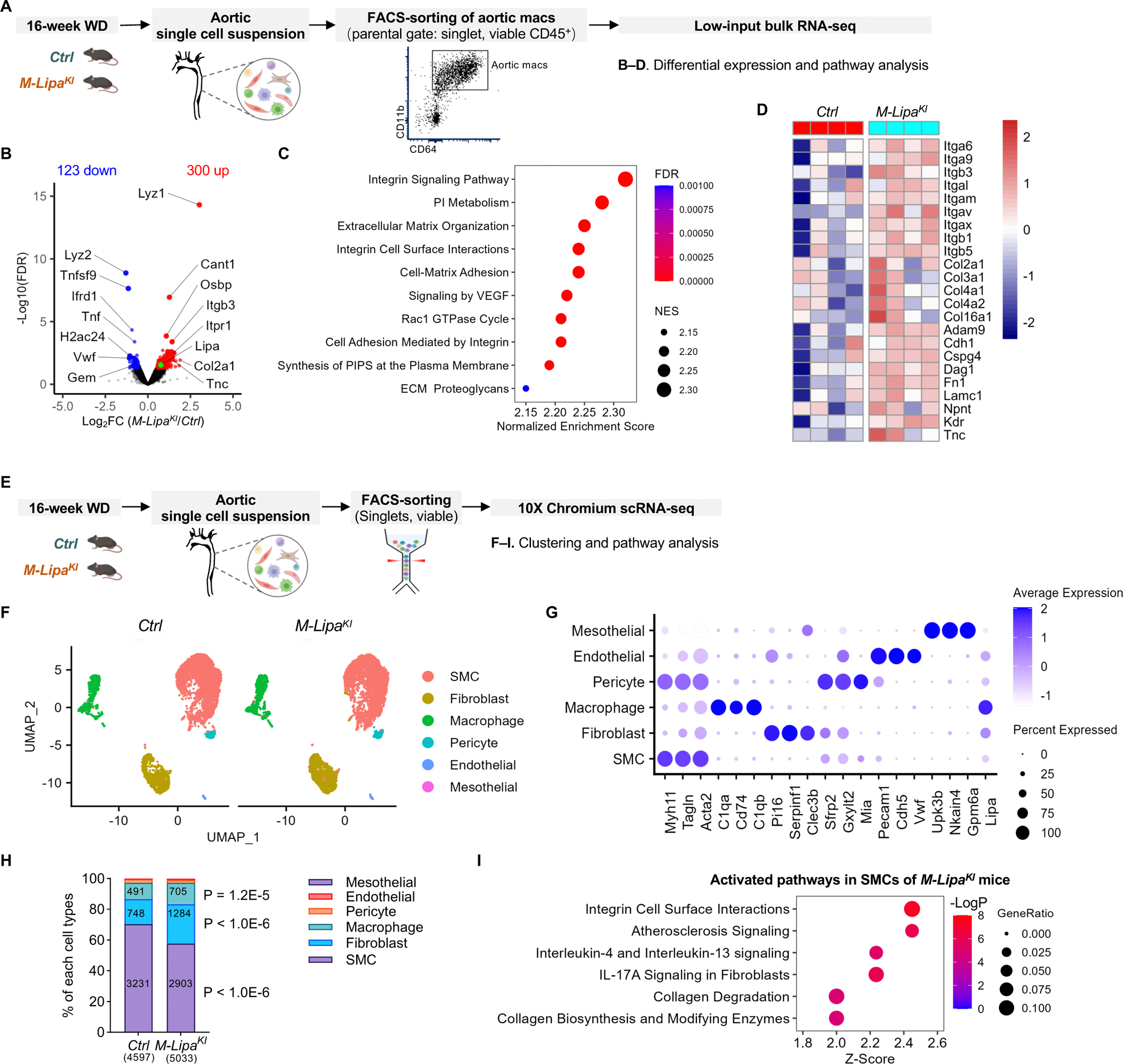
Methods: Post-genome-wide association study pipelines and molecular biology techniques, including expression quantitative trait loci analysis, Tri-HiC, luciferase assay, CRISPRi, allele-specific binding, motif analysis, and electrophoretic mobility shift assay, were used to link functional variants to target genes and define the direction of their regulatory effects in causal cell types. To determine how increased myeloid LIPA impacts atherosclerosis, myeloid-specific Lipa overexpression mice on an Ldlr-/- background were generated.
Results: Coronary artery disease-risk alleles in the LIPA locus increase LIPA expression and enzyme activity specifically in monocytes/macrophages by enhancing PU.1 binding to an intronic enhancer region that interacts with the LIPA promoter. Myeloid-specific Lipa overexpression in Ldlr-/- mice fed a western diet resulted in larger atherosclerotic lesions, accompanied by altered macrophage function, characterized by increased accumulation of lesional macrophages derived from circulating monocytes, reduced neutral lipid content, and up-regulation of integrin and extracellular matrix pathway genes.
Conclusions: The work establishes a direct causal link between LIPA-risk alleles and increased monocyte/macrophage LIPA that exacerbates atherosclerosis, bridging human functional genomic evidence to the mechanistic understanding of coronary artery disease.
Keywords: Atherosclerosis; Functional genomics; GWAS; Lysosomal acid lipase; Macrophage.
© The Author(s) 2025. Published by Oxford University Press on behalf of the European Society of Cardiology. All rights reserved. For commercial re-use, please contact reprints@oup.com for reprints and translation rights for reprints. All other permissions can be obtained through our RightsLink service via the Permissions link on the article page on our site—for further information please contact journals.permissions@oup.com.
Conflict of interest statement
Declarations
Disclosure of Interest
T.L. advises and has equity in Variant Bio and advised Goldfinch Bio and GSK. Other authors declare no disclosure of interest for this contribution.
Figures


References
MeSH terms
Substances
Grants and funding
- S10 OD019973/OD/NIH HHS/United States
- S10OD020056/GF/NIH HHS/United States
- S10 RR027050/RR/NCRR NIH HHS/United States
- R35 HL177389/HL/NHLBI NIH HHS/United States
- R01 MH106842/MH/NIMH NIH HHS/United States
- S10OD019973/GF/NIH HHS/United States
- R01 HL168174/HL/NHLBI NIH HHS/United States
- 20POST35130003 - FANG LI/AHA/American Heart Association-American Stroke Association/United States
- S10 OD020056/OD/NIH HHS/United States
- P30 DK063608/DK/NIDDK NIH HHS/United States
- P30CA013696/GF/NIH HHS/United States
- 23CDA1052177/AHA/American Heart Association-American Stroke Association/United States
- R00 HL130574/HL/NHLBI NIH HHS/United States
- F31 HG010580/HG/NHGRI NIH HHS/United States
- P30DK063608/GF/NIH HHS/United States
- P30 CA013696/CA/NCI NIH HHS/United States
- R01 HL151611/HL/NHLBI NIH HHS/United States
- S10RR027050/GF/NIH HHS/United States
- P01 HL172741/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
