

KRAS: the Achilles' heel of pancreas cancer biology
- PMID: 40829181
- PMCID: PMC12352898
- DOI: 10.1172/JCI191939
KRAS: the Achilles' heel of pancreas cancer biology
Abstract
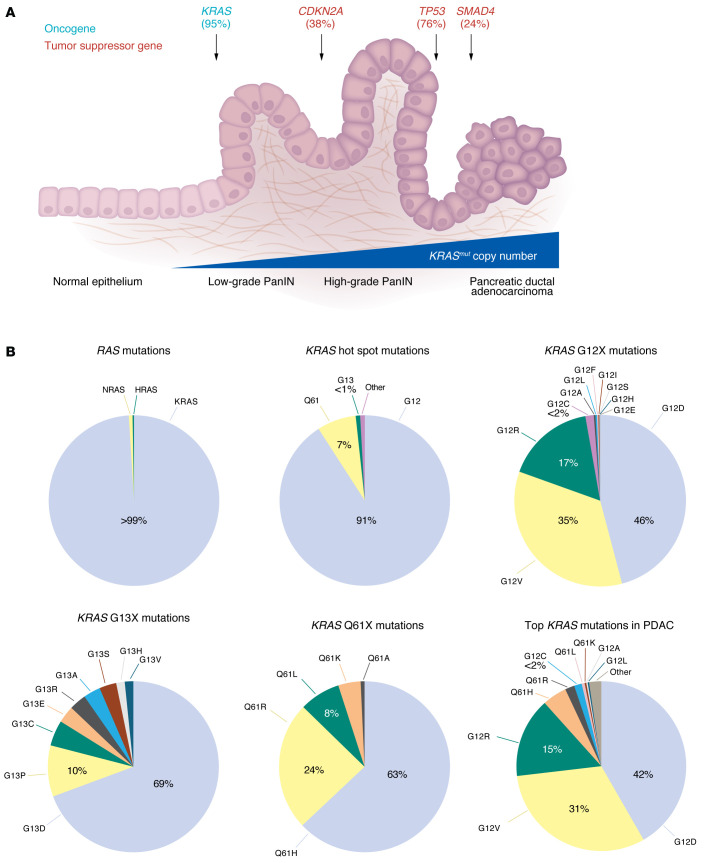
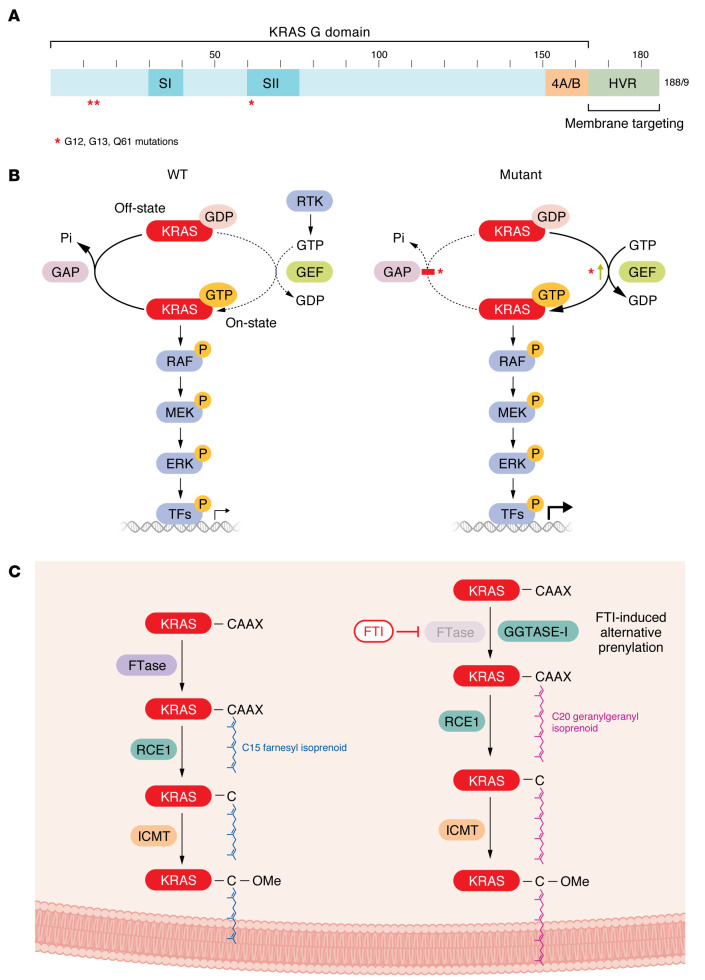
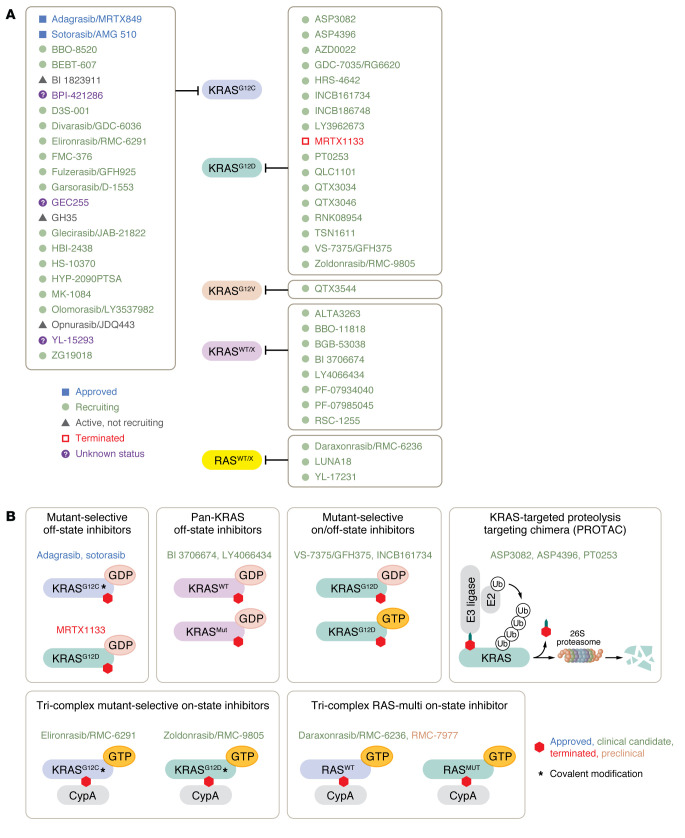
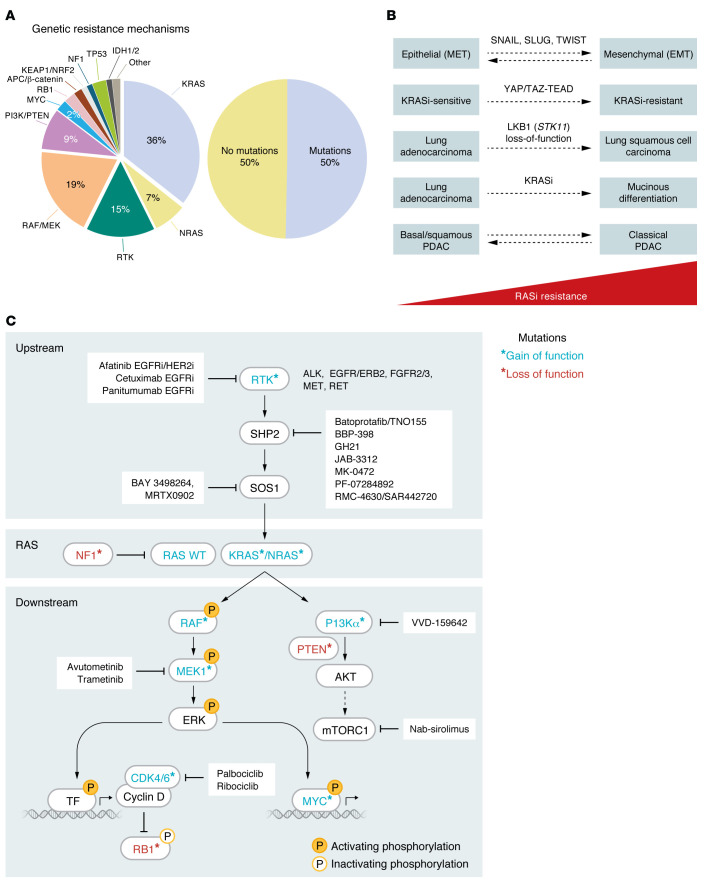
The genetic landscape of pancreatic ductal adenocarcinoma (PDAC) is well-established and dominated by four key genetic driver mutations. Mutational activation of the KRAS oncogene is the initiating genetic event, followed by genetic loss of function of the CDKN2A, TP53, and SMAD4 tumor suppressor genes. Disappointingly, this information has not been leveraged to develop clinically effective targeted therapies for PDAC treatment, where current standards of care remain cocktails of conventional cytotoxic drugs. Nearly all (~95%) PDAC harbors KRAS mutations, and experimental studies have validated the essential role of KRAS mutation in PDAC tumorigenic and metastatic growth. Identified in 1982 as the first gene shown to be aberrantly activated in human cancer, KRAS has been the focus of intensive drug discovery efforts. Widely considered "undruggable," KRAS has been the elephant in the room for PDAC treatment. This perception was shattered recently with the approval of two KRAS inhibitors for the treatment of KRASG12C-mutant lung and colorectal cancer, fueling hope that KRAS inhibitors will lead to a breakthrough in PDAC therapy. In this Review, we summarize the key role of aberrant KRAS signaling in the biology of pancreatic cancer; provide an overview of past, current, and emerging anti-KRAS treatment strategies; and discuss current challenges that limit the clinical efficacy of directly targeting KRAS for pancreatic cancer treatment.
Conflict of interest statement
Figures
Similar articles
-
ADT-1004: a first-in-class, oral pan-RAS inhibitor with robust antitumor activity in preclinical models of pancreatic ductal adenocarcinoma.Mol Cancer. 2025 Mar 13;24(1):76. doi: 10.1186/s12943-025-02288-9. Mol Cancer. 2025. PMID: 40082968 Free PMC article.
-
Drugging the 'undruggable' KRAS: breakthroughs, challenges, and opportunities in pancreatic cancer.Cancer Biol Med. 2025 Jul 7;22(7):762-88. doi: 10.20892/j.issn.2095-3941.2025.0122. Cancer Biol Med. 2025. PMID: 40624835 Free PMC article. Review.
-
Unveiling the intriguing relationship: oncogenic KRAS, morphological shifts, and mutational complexity in pancreatic mucinous cystic neoplasms.J Pathol. 2025 Apr;265(4):401-407. doi: 10.1002/path.6397. Epub 2025 Feb 5. J Pathol. 2025. PMID: 39906959 Free PMC article.
-
Pancreatic STAT5 activation promotes KrasG12D-induced and inflammation-induced acinar-to-ductal metaplasia and pancreatic cancer.Gut. 2024 Oct 7;73(11):1831-1843. doi: 10.1136/gutjnl-2024-332225. Gut. 2024. PMID: 38955401 Free PMC article.
-
Epidermal growth factor receptor (EGFR) inhibitors for metastatic colorectal cancer.Cochrane Database Syst Rev. 2017 Jun 27;6(6):CD007047. doi: 10.1002/14651858.CD007047.pub2. Cochrane Database Syst Rev. 2017. PMID: 28654140 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
