

MED12 mutation induces RTK inhibitor resistance in NSCLC via MEK/ERK pathway activation by inflammatory cytokines
- PMID: 40833497
- PMCID: PMC12367601
- DOI: 10.1007/s00018-025-05791-w
MED12 mutation induces RTK inhibitor resistance in NSCLC via MEK/ERK pathway activation by inflammatory cytokines
Abstract
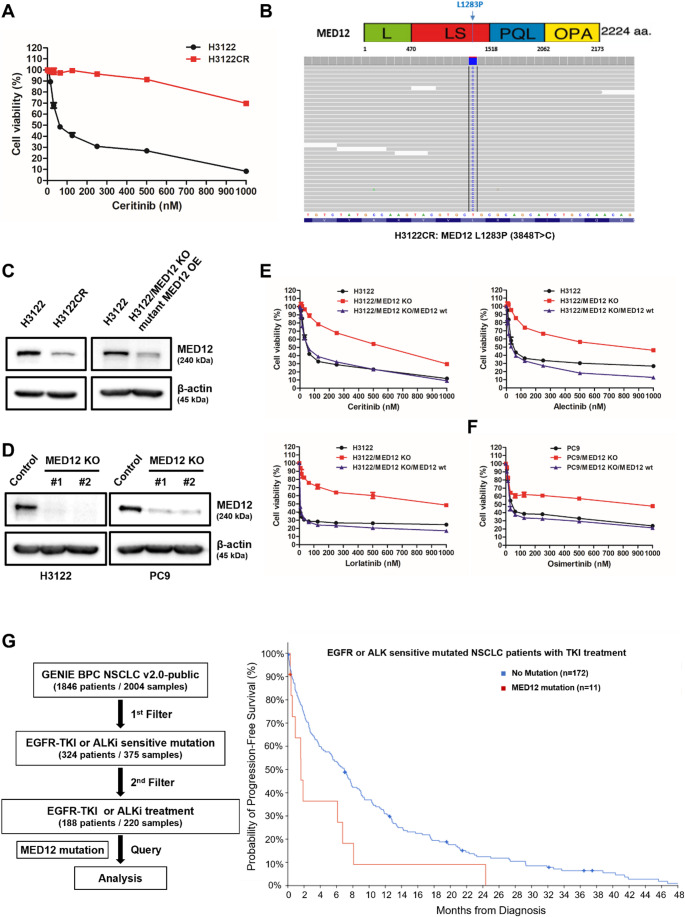
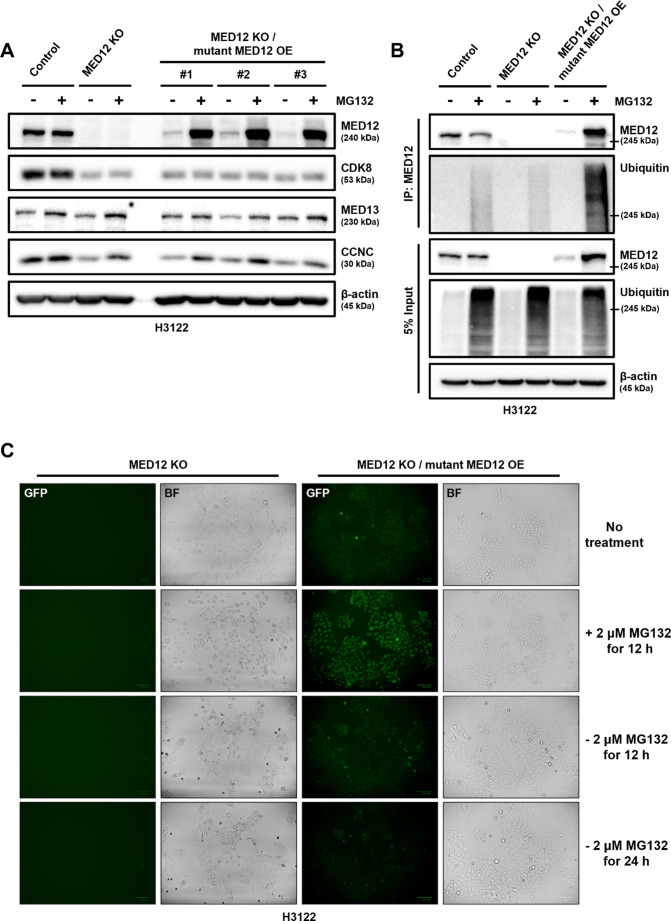
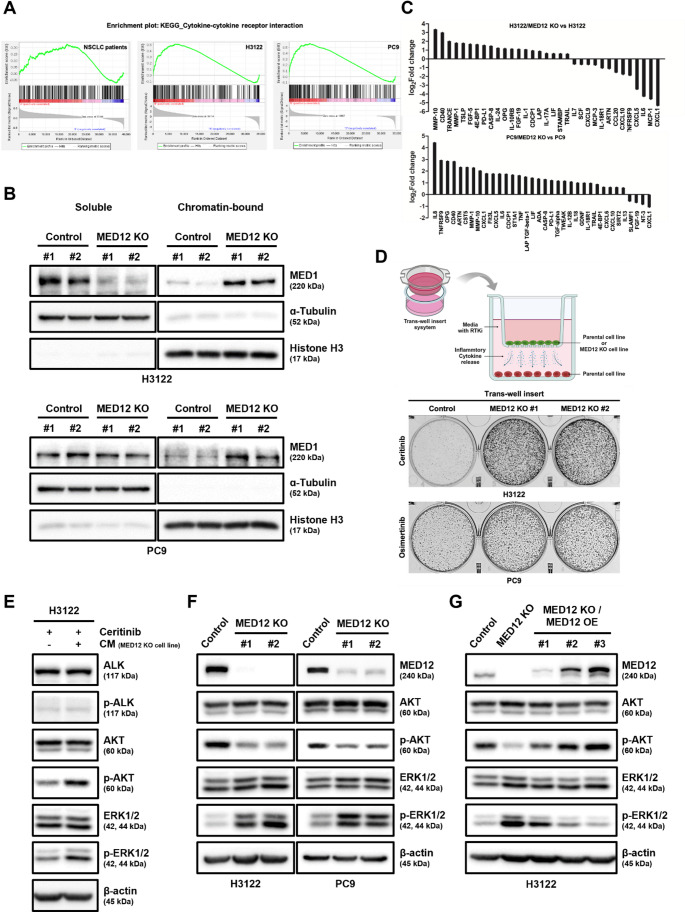
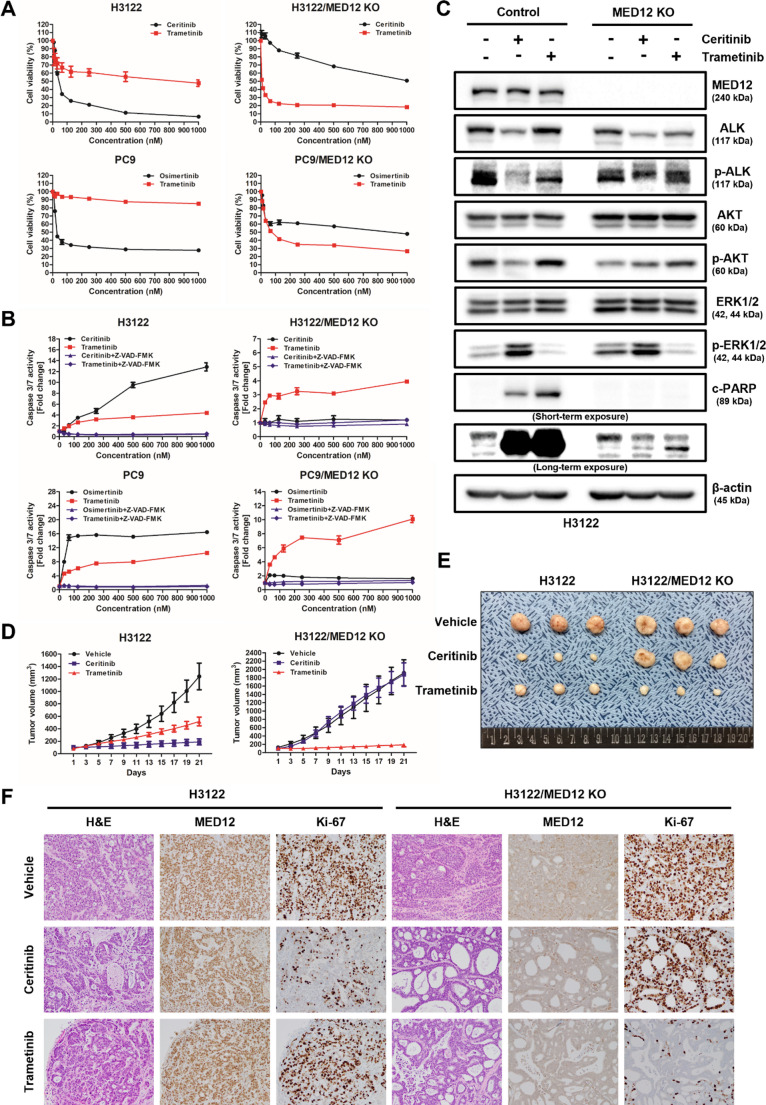
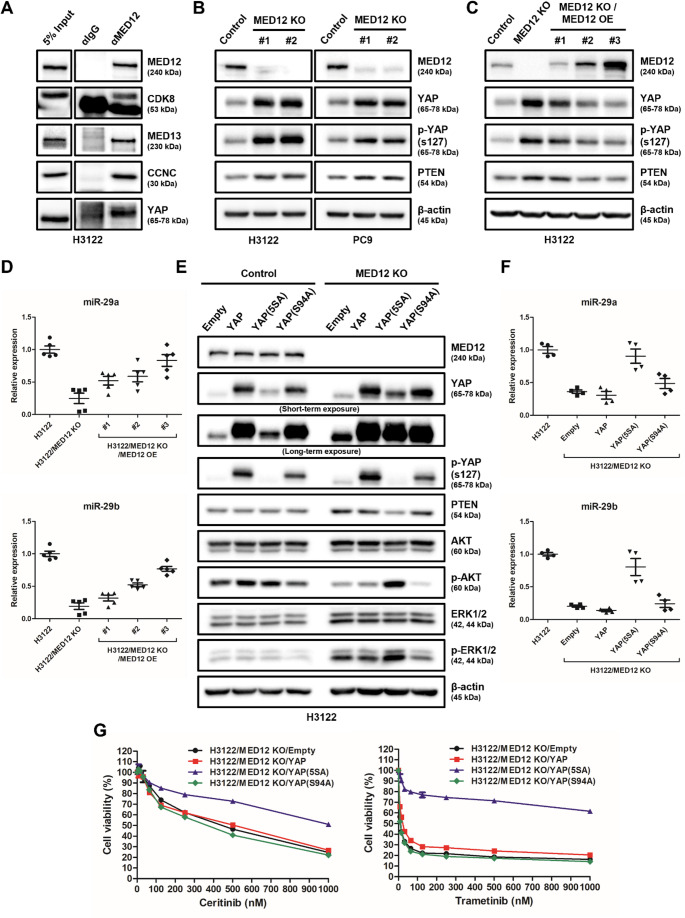
Non-small cell lung cancer (NSCLC) is frequently associated with mutations in receptor tyrosine kinases (RTKs), such as EGFR and ALK. While RTK inhibitors (RTKIs) have proven effective in treating patients with specific RTK mutations, the emergence of resistance to these therapies remains a significant clinical obstacle. As such, there is still an unmet need for the identification of new biomarkers that can predict resistance to RTK inhibitors in clinical use. In the present study, we demonstrate that MED12 mutations are a key driver of RTKi resistance in NSCLC cells. This resistance is mediated through the release of inflammatory cytokines triggered by MED12 degradation. Notably, we observed that of the two major downstream signaling pathways activated by inflammatory cytokines, only the MEK/ERK pathway was upregulated, while the PI3K/AKT pathway was unaffected in MED12 knock-out (KO) cells. The degradation of MED12 results in the dissociation of the MED12 complex, which subsequently leads to YAP phosphorylation. This phosphorylated YAP increases PTEN expression by inhibiting miR-29, thereby suppressing the PI3K/AKT signaling pathway. Importantly, treatment with trametinib, a MEK inhibitor, effectively suppressed tumor growth in MED12KO NSCLC cells and in xenograft models derived from these cells. These findings suggest that targeting the MEK/ERK signaling pathway, such as with trametinib, may provide a viable strategy to overcome RTKi resistance in MED12-mutant NSCLC. Furthermore, MED12 is identified as a crucial biomarker and potential therapeutic target for overcoming RTKi resistance.
Keywords: Drug resistance; MED12 mutation; MEK inhibitor; NSCLC; Receptor tyrosine kinase inhibitor.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval: All animal procedures were approved by the Asan Medical Centre Institutional Animal Care and Use Committee and Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines. Consent for publication: Not applicable. Competing interests: Dae Ho Lee declares honoraria from AstraZeneca, Boehringer-Ingelheim, Bristol-Myers Squibb, CJ Healthcare, ChongKunDang, Eli Lilly, Janssen, Merck, MSD, Mundipharma, Novartis, Ono, Pfizer, Roche, Samyang Biopharm and ST Cube. The other authors have no conflicts of interest to declare.
Figures

Similar articles
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Erlotinib or gefitinib for the treatment of relapsed platinum pretreated non-small cell lung cancer and ovarian cancer: a systematic review.Drug Resist Updat. 2011 Jun;14(3):177-90. doi: 10.1016/j.drup.2011.02.004. Epub 2011 Mar 24. Drug Resist Updat. 2011. PMID: 21435938
-
KRAS-mutant non-small cell lung cancer (NSCLC) therapy based on tepotinib and omeprazole combination.Cell Commun Signal. 2024 Jun 12;22(1):324. doi: 10.1186/s12964-024-01667-x. Cell Commun Signal. 2024. PMID: 38867255 Free PMC article.
-
MEK inhibitor trametinib combined with PI3K/mTOR inhibitor BEZ-235 as an effective strategy against NSCLC through impairment of glucose metabolism.Cell Signal. 2024 Dec;124:111415. doi: 10.1016/j.cellsig.2024.111415. Epub 2024 Sep 16. Cell Signal. 2024. PMID: 39293743
-
Systemic treatments for metastatic cutaneous melanoma.Cochrane Database Syst Rev. 2018 Feb 6;2(2):CD011123. doi: 10.1002/14651858.CD011123.pub2. Cochrane Database Syst Rev. 2018. PMID: 29405038 Free PMC article.
References
-
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S et al (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448:561–566 - PubMed
-
- Solomon B, Varella-Garcia M, Camidge DR (2009) ALK gene rearrangements: a new therapeutic target in a molecularly defined subset of non-small cell lung cancer. J Thorac Oncol 4:1450–1454 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
