

The genetic diversity and populational specificity of the human gut virome at single-nucleotide resolution
- PMID: 40836310
- PMCID: PMC12369081
- DOI: 10.1186/s40168-025-02185-9
The genetic diversity and populational specificity of the human gut virome at single-nucleotide resolution
Abstract
Background: Large-scale characterization of gut viral genomes provides strain-resolved insights into host-microbe interactions. However, existing viral genomes are mainly derived from Western populations, limiting our understanding of global gut viral diversity and functional variations necessary for personalized medicine and addressing regional health disparities.
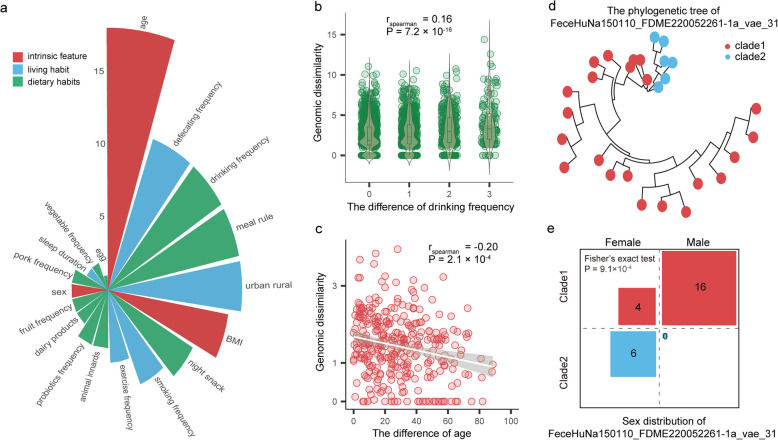
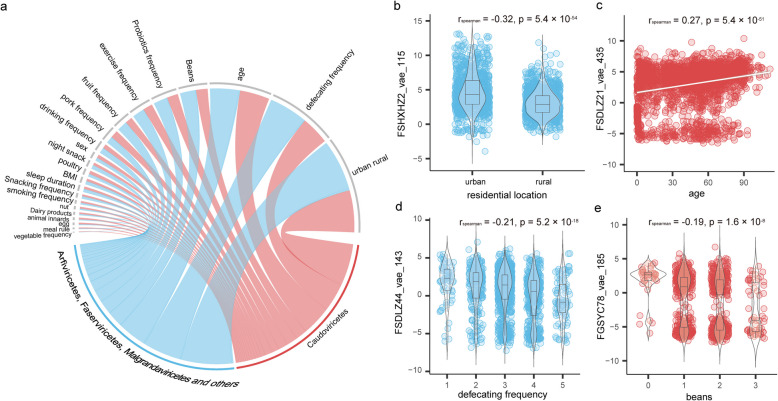
Results: Here, we introduce the Chinese Gut Viral Reference (CGVR) set, consisting of 120,568 viral genomes from 3234 deeply sequenced fecal samples collected nationwide, covering 72,751 viral operational taxonomic units (vOTUs), nearly 90% of which are likely absent from current databases. Analysis of single-nucleotide variations (SNVs) in 233 globally prevalent vOTUs revealed that 18.9% showed significant genetic stratification between Chinese and non-Chinese populations, potentially linked to bacterial infection susceptibility. The predicted bacterial hosts of population-stratified viruses exhibit distinct genetic components associated with health-related functions, including multidrug resistance. Additionally, viral strain diversity at the SNV level correlated with human phenotypic traits, such as age and gastrointestinal issues like constipation. Our analysis also indicates that the human gut bacteriome is specifically shaped by the virome, which mediates associations with human phenotypic traits. Video Abstract CONCLUSIONS: Our analysis underscores the unique genetic makeup of the gut virome across populations and emphasizes the importance of recognizing gut viral genetic heterogeneity for deeper insights into regional health implications.
Keywords: Bioinformatics; Cohort study; Genomic epidemiology; Metagenomics; Virome.
© 2025. The Author(s).
Conflict of interest statement
Declarations: Ethics approval and consent to participate. The study was approved by the Ethics Review Board of Jiangnan University (reference number JNU20220901IRB01) and included 3,234 participants from 30 provinces across Mainland China. Informed consent was obtained from all participants prior to sample collection. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures





Similar articles
-
Biases and complementarity in gut viromes obtained from bulk and virus-like particle-enriched metagenomic sequencing.Microbiol Spectr. 2025 Aug 5;13(8):e0001325. doi: 10.1128/spectrum.00013-25. Epub 2025 Jul 2. Microbiol Spectr. 2025. PMID: 40600714 Free PMC article.
-
Genetic characterization of the marmot gut virome in high-altitude Qinghai Province and identification of novel viruses with zoonotic potential.mSphere. 2025 Aug 26;10(8):e0029725. doi: 10.1128/msphere.00297-25. Epub 2025 Jul 31. mSphere. 2025. PMID: 40742122 Free PMC article.
-
Impact of diet in shaping gut virome of grain-fed and grass-fed beef cattle revealed by a comparative metagenomic study.Microbiome. 2025 Aug 23;13(1):190. doi: 10.1186/s40168-025-02163-1. Microbiome. 2025. PMID: 40849482 Free PMC article.
-
Analysis of the overall development trends and hotspots in the research field of the human gut virome.Virol J. 2025 Jul 5;22(1):224. doi: 10.1186/s12985-025-02856-x. Virol J. 2025. PMID: 40618116 Free PMC article. Review.
-
Type 1 Diabetes: A Guide to Autoimmune Mechanisms for Clinicians.Diabetes Obes Metab. 2025 Aug;27 Suppl 6(Suppl 6):40-56. doi: 10.1111/dom.16460. Epub 2025 May 15. Diabetes Obes Metab. 2025. PMID: 40375390 Free PMC article. Review.
References
-
- Yang K, Niu J, Zuo T, Sun Y, Xu Z, Tang W, Liu Q, Zhang J, Ng EKW, Wong SKH, et al. Alterations in the gut virome in obesity and type 2 diabetes mellitus. Gastroenterology. 2021;161(4):1257-1269.e1213. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
