

Exploiting viral infection/vaccination to focus high-affinity T cell populations into tumors using oncolytic viro-immunotherapy
- PMID: 40842156
- PMCID: PMC12502006
- DOI: 10.1016/j.ymthe.2025.08.023
Exploiting viral infection/vaccination to focus high-affinity T cell populations into tumors using oncolytic viro-immunotherapy
Abstract
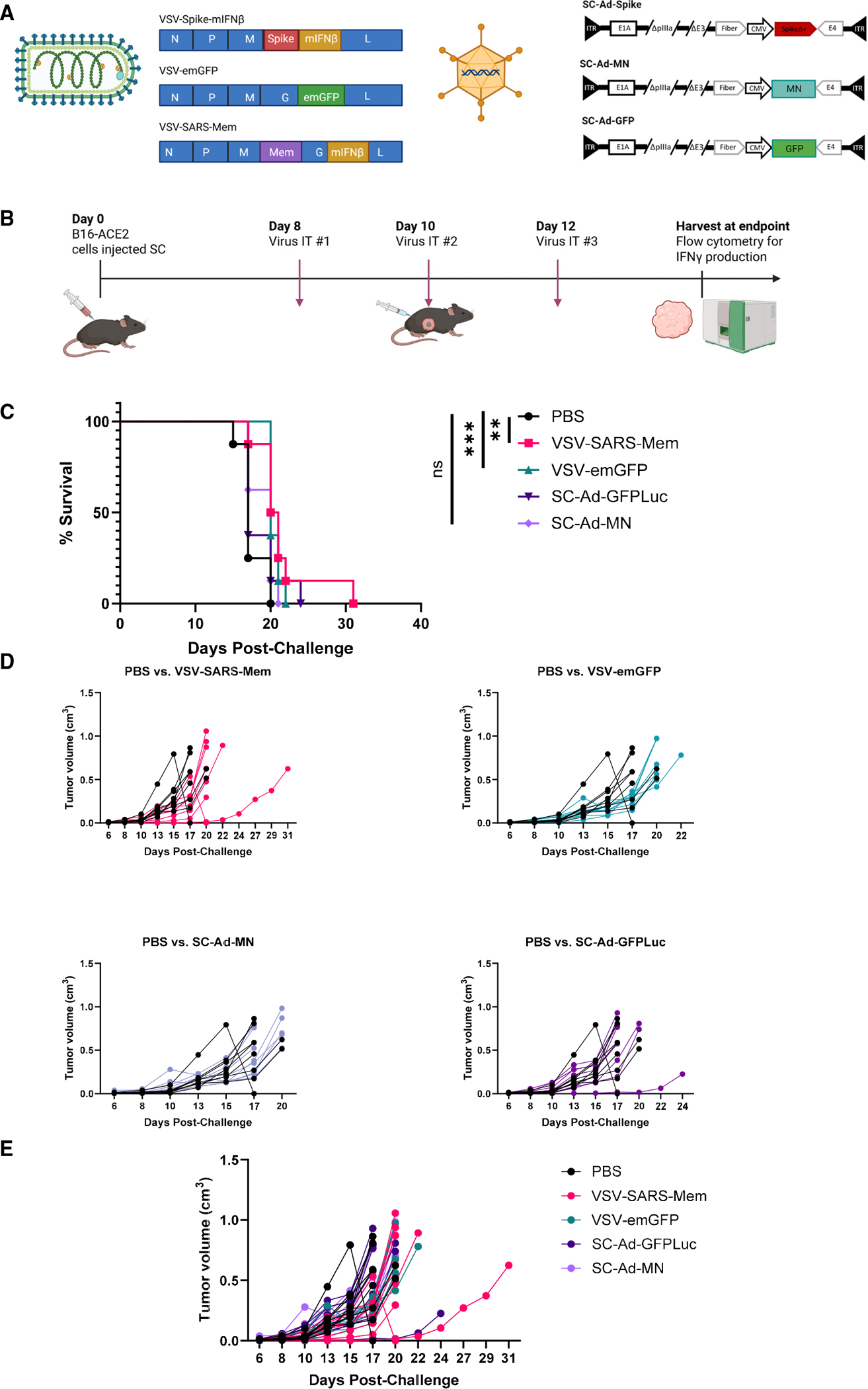
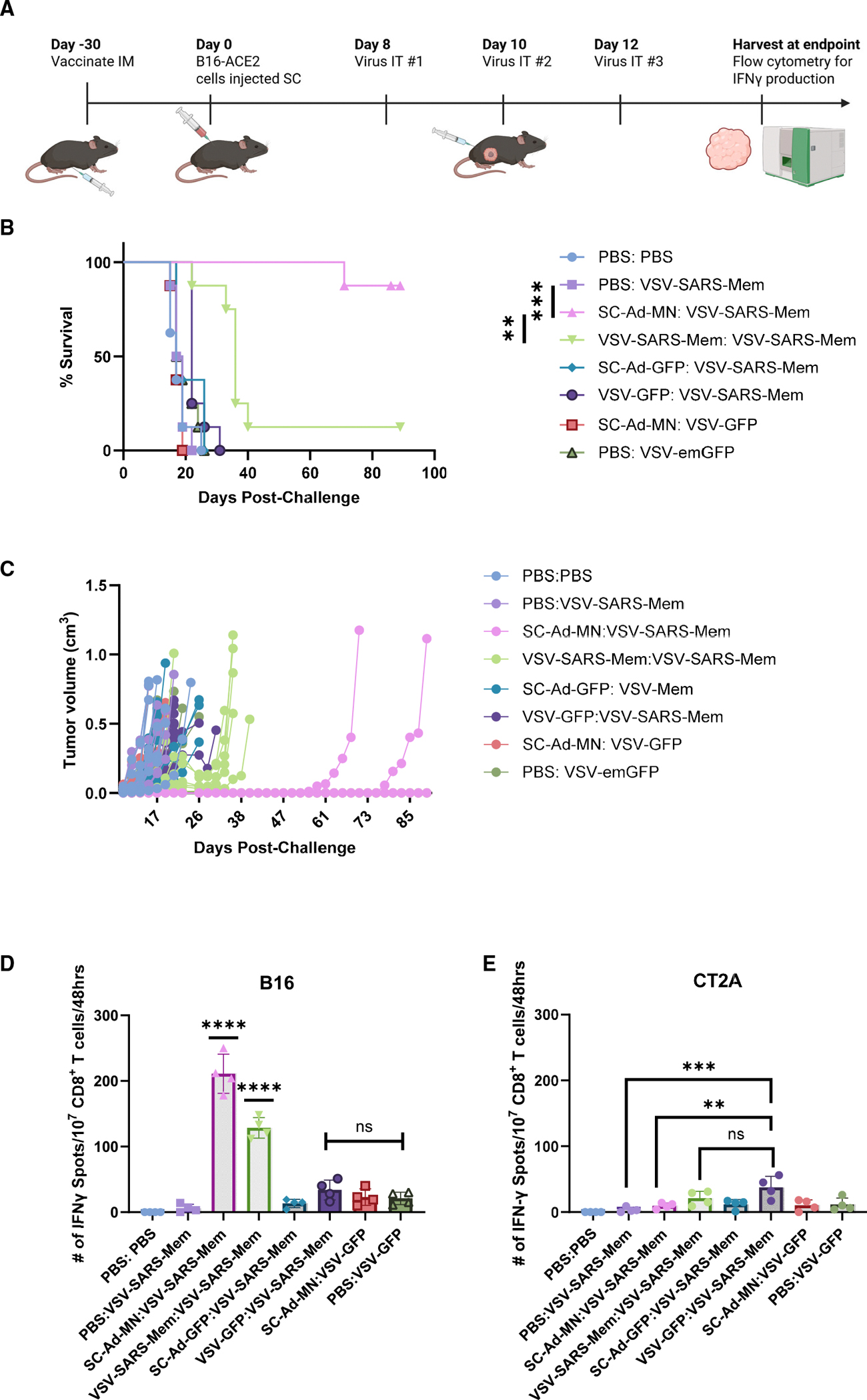
Immune tolerance restricts the number of T cells with significant affinity for self-tumor-associated antigens (TAAs), thereby limiting successful cancer immunotherapy through an inability to generate populations of high-affinity anti-tumor T cells. In contrast, viral infection/vaccination primes and expands high-affinity effector and memory T cells against viral antigens. We show here that it is possible to exploit population-wide preexisting, anti-viral memory recall responses against SARS-CoV-2 antigens to focus a high-affinity, immunodominant T cell response into tumors by oncolytic virus (OV)-mediated or chimeric antigen receptor (CAR)-mediated delivery of viral antigens that are not themselves related to TAAs. Heterologous prime and OV/boost led to CD8+ T cell-dependent tumor cures using either SARS-CoV-2 Mem or Spike (S) proteins as vaccinating/tumor-focusing T cell targets, associated with epitope spreading against TAAs. We also show that CAR-T cells carry SARS-CoV-2 antigen-expressing vectors systemically to tumors even in pre-immune mice. Finally, S-specific CAR-T cells could be boosted in vivo with S protein vaccines to enhance anti-tumor activity and persistence. Thus, where high affinity anti-tumor T cells are not available, boosting preexisting infection- or vaccination-induced T cell populations within tumors using OV-mediated immunogen delivery provides a therapeutically valuable alternative.
Keywords: SARS-CoV-2; cancer immunotherapy; oncolytic viruses; single cycle adenovirus; tumor antigens; vaccines; vesicular stomatitis virus; virus T cell memory.
Copyright © 2025 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures




References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
