

NMDA-Type Glutamate Receptor Activation Promotes Ischemic Arrhythmias by Targeting the AKT1-TBX3-Nav1.5 Axis
- PMID: 40844176
- PMCID: PMC12372452
- DOI: 10.1111/apha.70085
NMDA-Type Glutamate Receptor Activation Promotes Ischemic Arrhythmias by Targeting the AKT1-TBX3-Nav1.5 Axis
Abstract
Aim: The aim of this study is to determine the possible role of N-methyl-D-aspartate receptor (NMDAR) dysregulation in the ischemic electrical remodeling observed in patients with myocardial infarction (MI) and elucidate the underlying mechanisms.
Methods: Human heart tissue was obtained from the border of the infarct and remote zones of patients with ischemic heart disease, and mouse heart tissue was obtained from the peri-infarct zone. NMDAR expression was detected using immunofluorescence (IF) and Western blotting (WB). Spontaneous ventricular arrhythmias (VAs) in mice were detected using electrocardiogram backpacks. Electrical remodeling post-MI was detected using patch clamp recordings, quantitative real-time polymerase chain reactions, IF, and WB. Mechanistic studies were performed using bioinformatic analysis, plasmid and small interfering RNA transfection, lentiviral packaging, and site-directed mutagenesis.
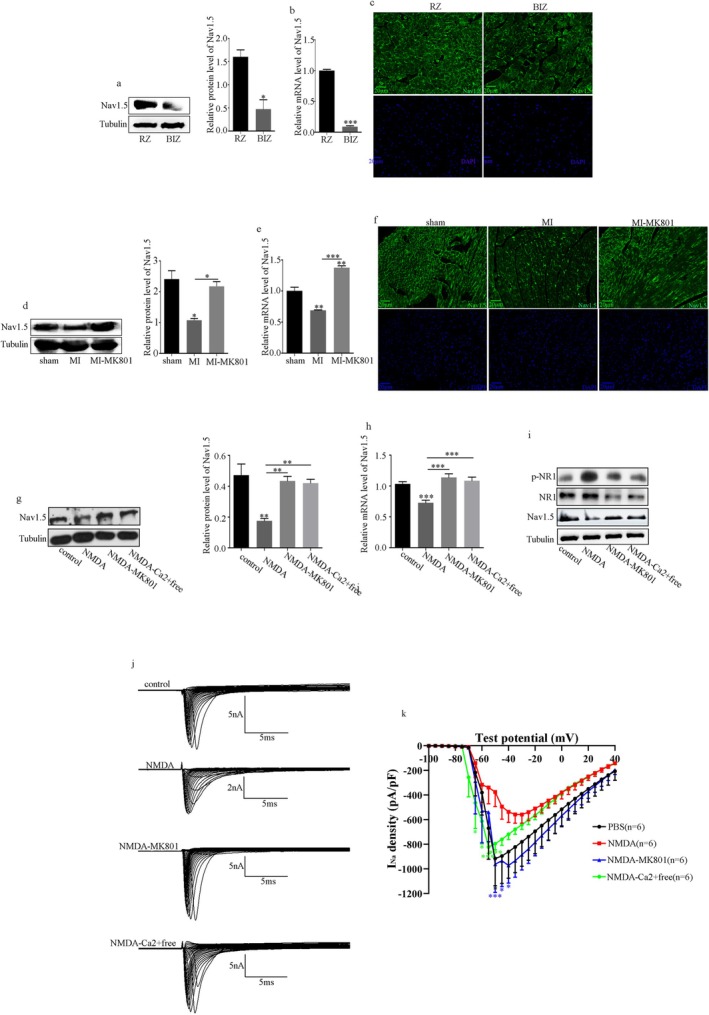
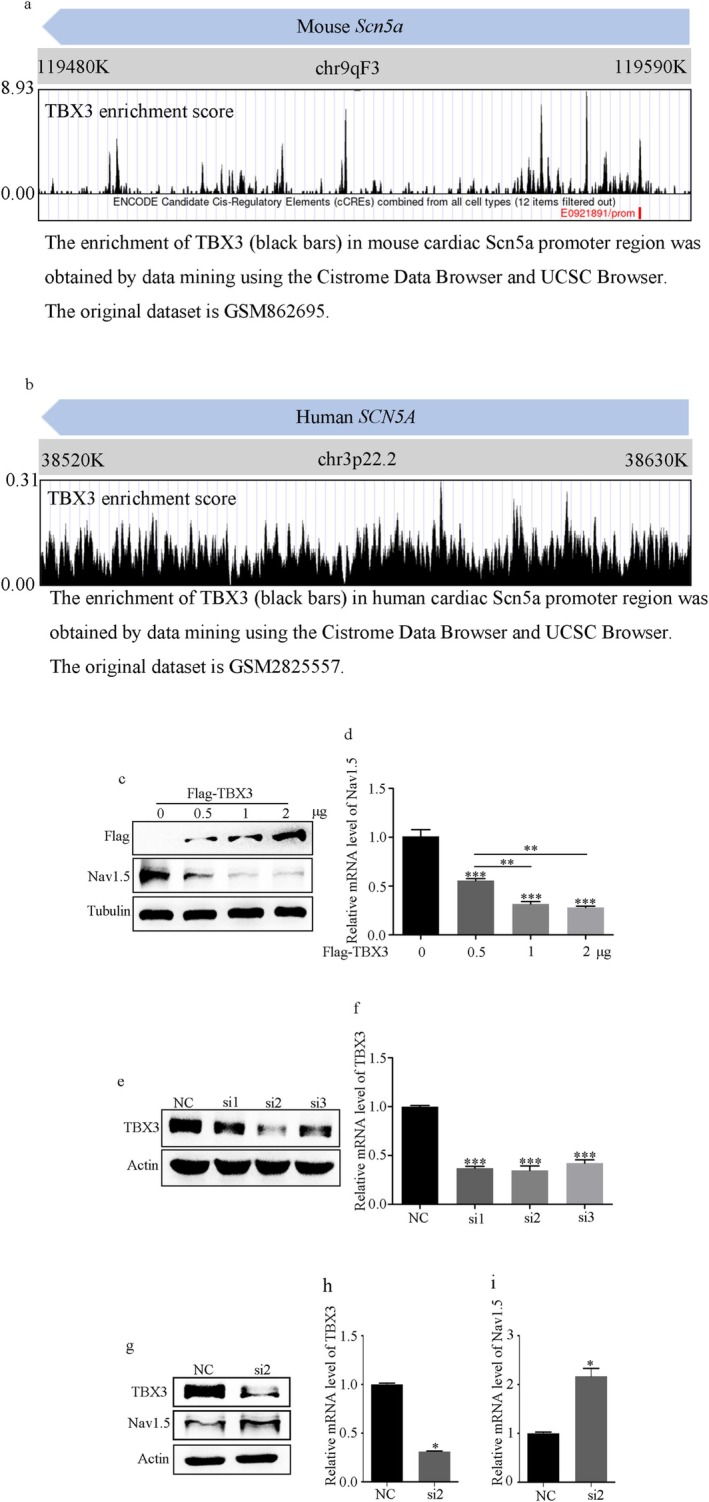
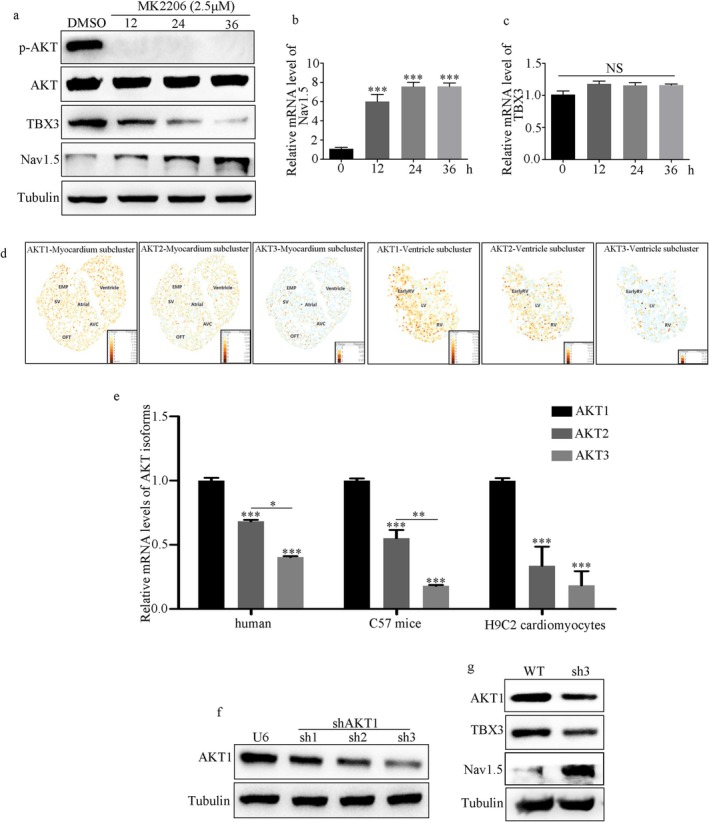
Results: NMDAR is highly expressed in patients with ischemic heart disease and mice with MI. NMDAR inhibition reduces the occurrence of VAs. Mechanistically, NMDAR activation promotes electrophysiological remodeling, as characterized by decreased Nav1.5, Kv11.1, Kv4.2, Kv7.1, Kir2.1, and Cav1.2 expression in patients with ischemic heart disease and mice with MI and rescues these ion channels dysregulation in mice with MI to varying degrees by NMDAR inhibition. Decreased Nav1.5 expression and inward sodium current density were attenuated by NMDAR inhibition in primary rat cardiomyocytes. Moreover, NMDAR activation upregulates T-Box Transcription Factor 3 (TBX3) post-translationally, further downregulating Nav1.5 transcriptionally. Furthermore, AKT1 is the predominant isoform in the ventricular myocardium upstream of TBX3 and mediates NMDAR-induced TBX3 upregulation in cardiomyocytes.
Conclusion: NMDAR activation contributes to MI-induced VAs by regulating the AKT1-TBX3-Nav1.5 axis, providing novel therapeutic strategies for treating ischemic arrhythmias.
Keywords: NMDAR; electrophysiological remodeling; myocardial infarction; pseudo‐phosphorylation; ventricular arrhythmias.
© 2025 The Author(s). Acta Physiologica published by John Wiley & Sons Ltd on behalf of Scandinavian Physiological Society.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures




References
-
- Myerburg R. J. and Junttila M. J., “Sudden Cardiac Death Caused by Coronary Heart Disease,” Circulation 125, no. 8 (2012): 1043–1052. - PubMed
-
- Rouleau J. L., Talajic M., Sussex B., et al., “Myocardial Infarction Patients in the 1990s—Their Risk Factors, Stratification and Survival in Canada: The Canadian Assessment of Myocardial Infarction (CAMI) Study,” Journal of the American College of Cardiology 27, no. 5 (1996): 1119–1127. - PubMed
-
- Wasson S., Reddy H. K., and Dohrmann M. L., “Current Perspectives of Electrical Remodeling and Its Therapeutic Implications,” Journal of Cardiovascular Pharmacology and Therapeutics 9, no. 2 (2004): 129–144. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
