

AKR1B10 dictates c-Myc stability to suppress colorectal cancer metastasis via PP2A nitration
- PMID: 40845116
- PMCID: PMC12372902
- DOI: 10.1126/sciadv.adv6937
AKR1B10 dictates c-Myc stability to suppress colorectal cancer metastasis via PP2A nitration
Abstract
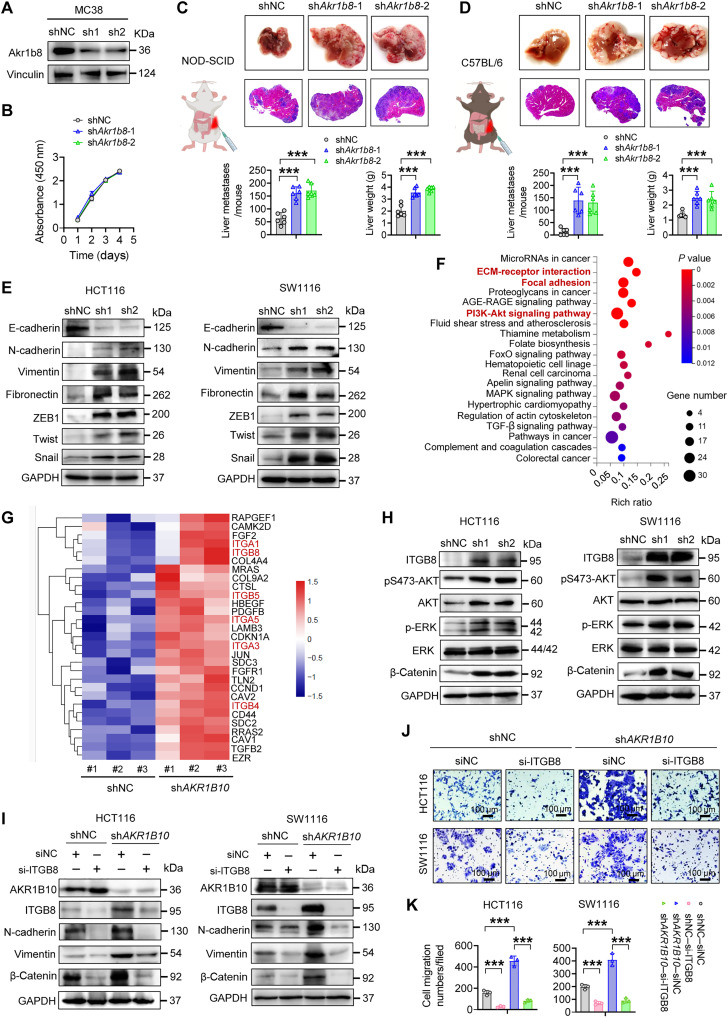
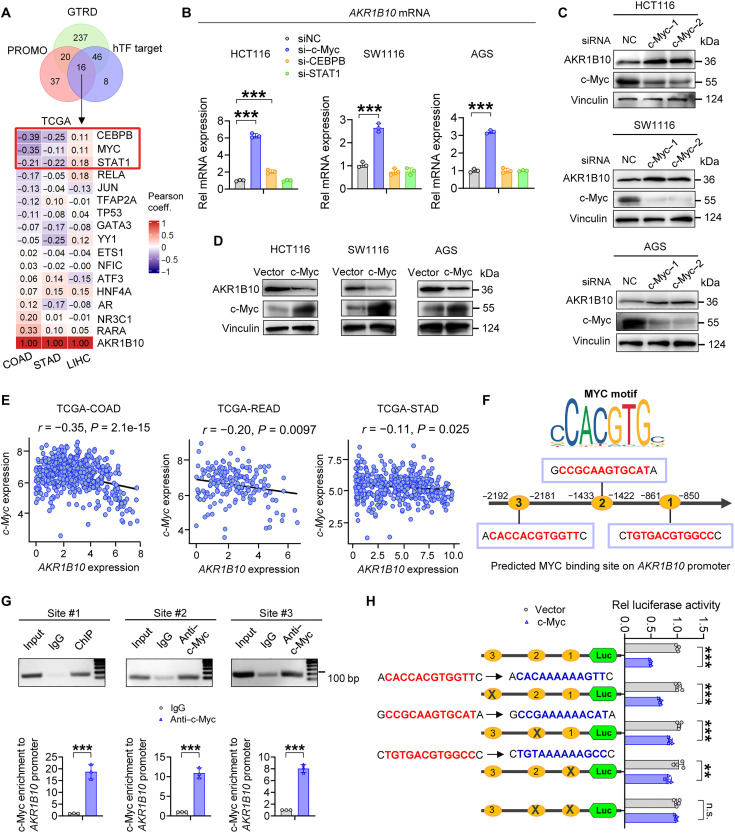
Metabolic enzymes, critical for cellular homeostasis, are frequently co-opted in a disease-specific manner to drive cancer progression. Here, we identify aldo-keto reductase family 1 member B10 (AKR1B10), down-regulated in gastrointestinal cancers, as a pivotal metastasis suppressor correlating with improved colorectal cancer (CRC) prognosis. Mechanistically, AKR1B10 activates protein phosphatase 2A (PP2A) by preventing redox-regulated nitration of its B56α subunit, preserving holoenzyme assembly and enabling c-Myc dephosphorylation at serine-62. Loss of AKR1B10 disrupts this pathway, stabilizing c-Myc, which drives integrin signaling and metastatic dissemination in CRC. We further demonstrate that lysine-125 of AKR1B10 is essential for its interaction with PP2A-Cα and B56α nitration, thereby attenuating CRC metastatic aggressiveness. Pharmacological restoration of PP2A activity effectively mitigates metastasis associated with AKR1B10 loss. In addition, c-Myc transcriptionally represses AKR1B10, establishing a feedback loop that sustains its down-regulation and enhances metastatic progression. This study uncovers an antimetastatic mechanism involving AKR1B10-mediated PP2A activation and highlights its potential as a biomarker and therapeutic target.
Figures







Similar articles
-
Aldo-keto Reductase 1B10 (AKR1B10) Suppresses Sensitivity of Ferroptosis in TNBC by Activating the AKT/GSK3β/Nrf2/GPX4 Axis.Front Biosci (Landmark Ed). 2025 Jun 27;30(6):36615. doi: 10.31083/FBL36615. Front Biosci (Landmark Ed). 2025. PMID: 40613296
-
Dual compartment utility of BRET-based biosensors for PPP2R5A/B56α, a cancer-associated B regulatory subunit of PP2A.Biotechniques. 2025 Apr;77(4):153-163. doi: 10.1080/07366205.2025.2523093. Epub 2025 Jun 27. Biotechniques. 2025. PMID: 40579746
-
Sialylated IgG-activated integrin β4-Src-Erk axis stabilizes c-Myc in a p300 lysine acetyltransferase-dependent manner to promote colorectal cancer liver metastasis.Neoplasia. 2025 Mar;61:101140. doi: 10.1016/j.neo.2025.101140. Epub 2025 Feb 25. Neoplasia. 2025. PMID: 40010102 Free PMC article.
-
Role of Mesenchymal Markers in Colorectal Cancer Metastasis.Mol Biol Rep. 2025 Jul 4;52(1):673. doi: 10.1007/s11033-025-10745-3. Mol Biol Rep. 2025. PMID: 40613936 Review.
-
Probing the Depths of Molecular Complexity: STAT3 as a Key Architect in Colorectal Cancer Pathogenesis.Curr Gene Ther. 2025;25(4):433-452. doi: 10.2174/0115665232336447241010094744. Curr Gene Ther. 2025. PMID: 39473261 Review.
References
-
- Siegel R. L., Miller K. D., Fuchs H. E., Jemal A., Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022). - PubMed
-
- Cañellas-Socias A., Sancho E., Batlle E., Mechanisms of metastatic colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 21, 609–625 (2024). - PubMed
-
- Biller L. H., Schrag D., Diagnosis and treatment of metastatic colorectal cancer: A review. JAMA 325, 669–685 (2021). - PubMed
-
- Arnold M., Sierra M. S., Laversanne M., Soerjomataram I., Jemal A., Bray F., Global patterns and trends in colorectal cancer incidence and mortality. Gut 66, 683–691 (2017). - PubMed
-
- Chowdhury D., Keogh M.-C., Ishii H., Peterson C. L., Buratowski S., Lieberman J., γ-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol. Cell 20, 801–809 (2005). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
