

Crosstalk Between Microbiome and Ferroptosis in Diseases: From Mechanism to Therapy
- PMID: 40846688
- PMCID: PMC12373584
- DOI: 10.1002/cph4.70042
Crosstalk Between Microbiome and Ferroptosis in Diseases: From Mechanism to Therapy
Abstract
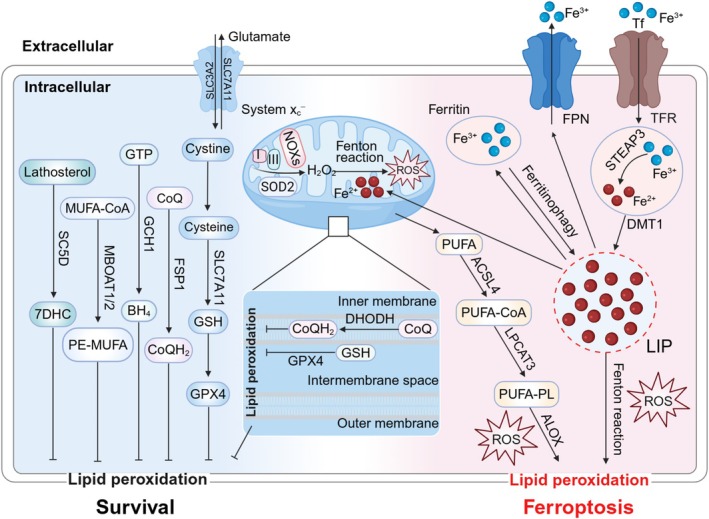
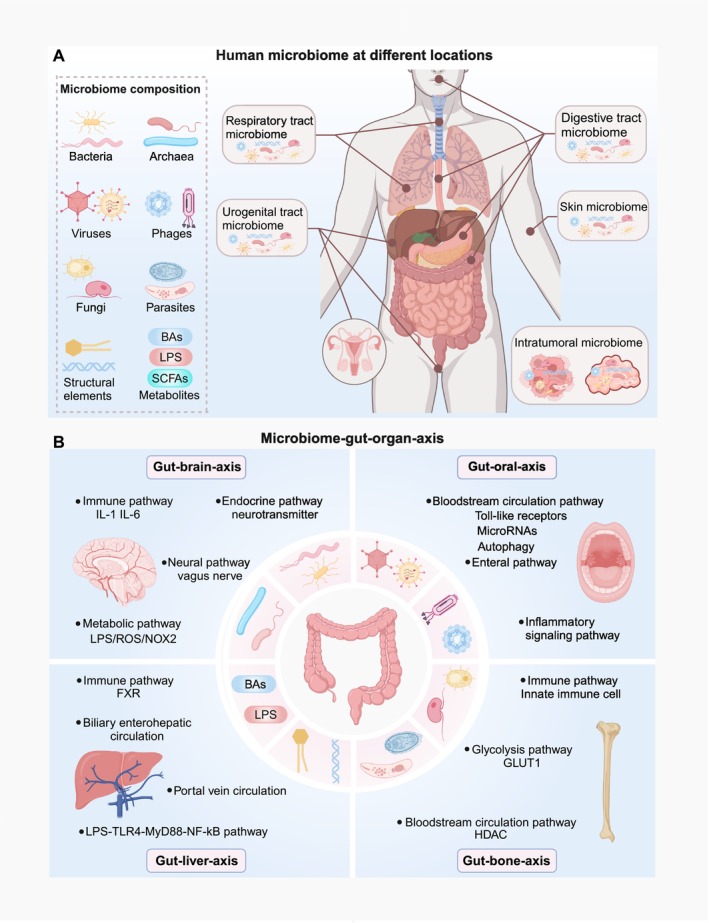
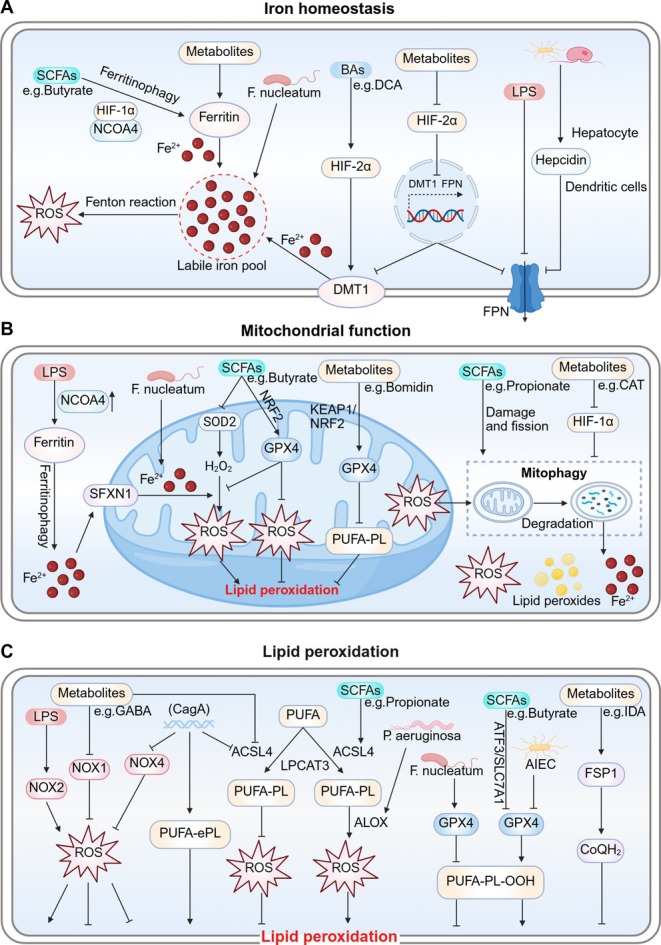
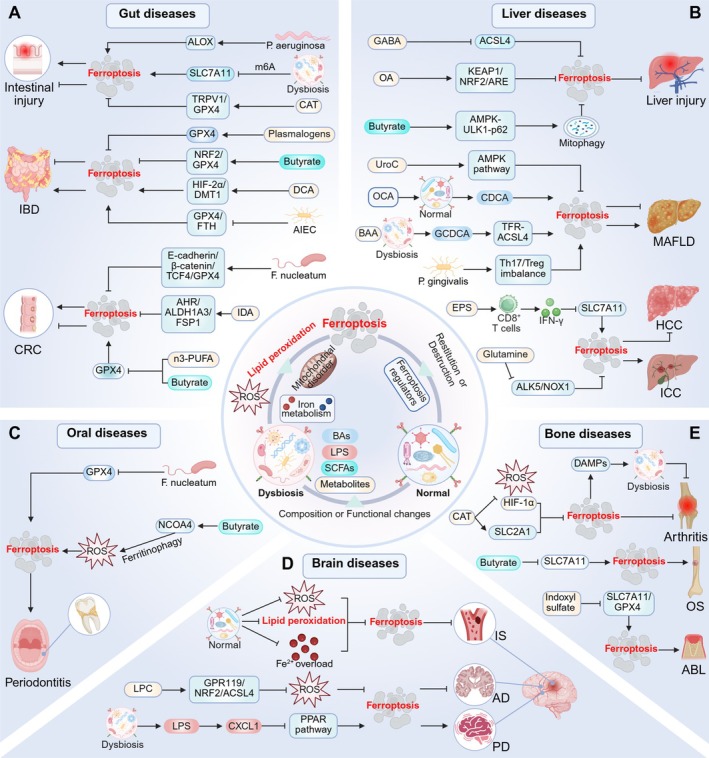
The human microbiome is a unique organ and maintains host immunomodulation and nutrient metabolism. Structural and functional microbiome alterations are commonly known as dysbiosis, which is strongly associated with disease progression. Ferroptosis is a novel iron-dependent cell death mode characterized by intracellular iron accumulation, increased reactive oxygen species (ROS), and lipid peroxidation (LPO). Importantly, the complex crosstalk between the microbiome and ferroptosis in disease has attracted considerable research attention. The microbiome influences ferroptosis by regulating host iron homeostasis, mitochondrial metabolism, and LPO, among many other pathways. Thus, the in-depth analysis of microbiome-ferroptosis crosstalk and associated mechanisms could provide new strategies to treat human diseases. Therefore, understanding this crosstalk is critical. Here, we systematically explore the associations between gut microbiome and ferroptosis across multiple diseases. We show that the oral microbiome also influences disease progression by regulating ferroptosis. Furthermore, we provide a potential for certain disease therapies by targeting the crosstalk between the microbiome and ferroptosis.
Keywords: fecal microbiota transplantation; ferroptosis; gut‐organ‐axis; microbiome; probiotics.
© 2025 The Author(s). Comprehensive Physiology published by Wiley Periodicals LLC on behalf of American Physiological Society.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Ferroptosis and gut microbiota: A new horizon in alcohol-associated liver disease management.Cell Mol Life Sci. 2025 Jul 19;82(1):282. doi: 10.1007/s00018-025-05815-5. Cell Mol Life Sci. 2025. PMID: 40682668 Free PMC article. Review.
-
Analyzing the role of ferroptosis in ribosome-related bone marrow failure disorders: From pathophysiology to potential pharmacological exploitation.IUBMB Life. 2024 Dec;76(12):1011-1034. doi: 10.1002/iub.2897. Epub 2024 Jul 25. IUBMB Life. 2024. PMID: 39052023 Free PMC article. Review.
-
Uncovering the role of ferroptosis in Bietti crystalline dystrophy and potential therapeutic strategies.Cell Commun Signal. 2024 Jul 11;22(1):359. doi: 10.1186/s12964-024-01710-x. Cell Commun Signal. 2024. PMID: 38992691 Free PMC article.
-
BNIP3-mediated mitophagy attenuates hypoxic-ischemic brain damage in neonatal rats by inhibiting ferroptosis through P62-KEAP1-NRF2 pathway activation to maintain iron and redox homeostasis.Acta Pharmacol Sin. 2025 Jan;46(1):33-51. doi: 10.1038/s41401-024-01365-x. Epub 2024 Aug 23. Acta Pharmacol Sin. 2025. PMID: 39179868
-
The Crosstalk Between Ferritinophagy and Ferroptosis in Ischemic Stroke: Regulatory Mechanisms and Therapeutic Implications.Cell Mol Neurobiol. 2025 Jul 20;45(1):73. doi: 10.1007/s10571-025-01593-7. Cell Mol Neurobiol. 2025. PMID: 40684405 Free PMC article. Review.
References
-
- Abed, J. , Maalouf N., Manson A. L., et al. 2020. “Colon Cancer‐Associated <styled-content style="fixed-case"> Fusobacterium nucleatum </styled-content> May Originate From the Oral Cavity and Reach Colon Tumors via the Circulatory System.” Frontiers in Cellular and Infection Microbiology 10: 400. - PMC - PubMed
-
- Ahlqvist, K. J. , Leoncini S., Pecorelli A., et al. 2015. “MtDNA Mutagenesis Impairs Elimination of Mitochondria During Erythroid Maturation Leading to Enhanced Erythrocyte Destruction.” Nature Communications 6: 6494. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
