

Tamoxifen induces PI3K activation in uterine cancer
- PMID: 40846762
- PMCID: PMC12425819
- DOI: 10.1038/s41588-025-02308-w
Tamoxifen induces PI3K activation in uterine cancer
Abstract
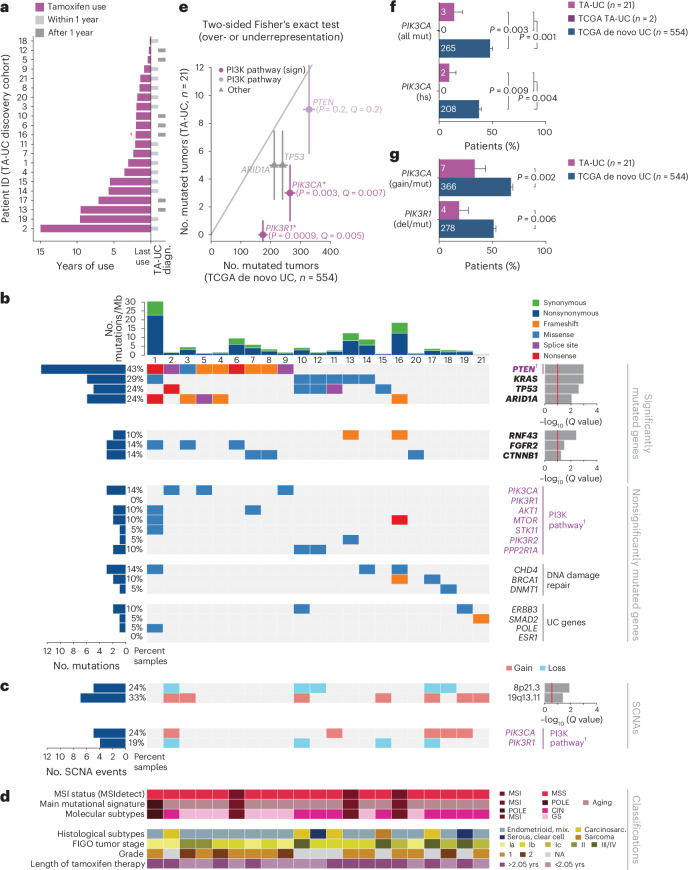
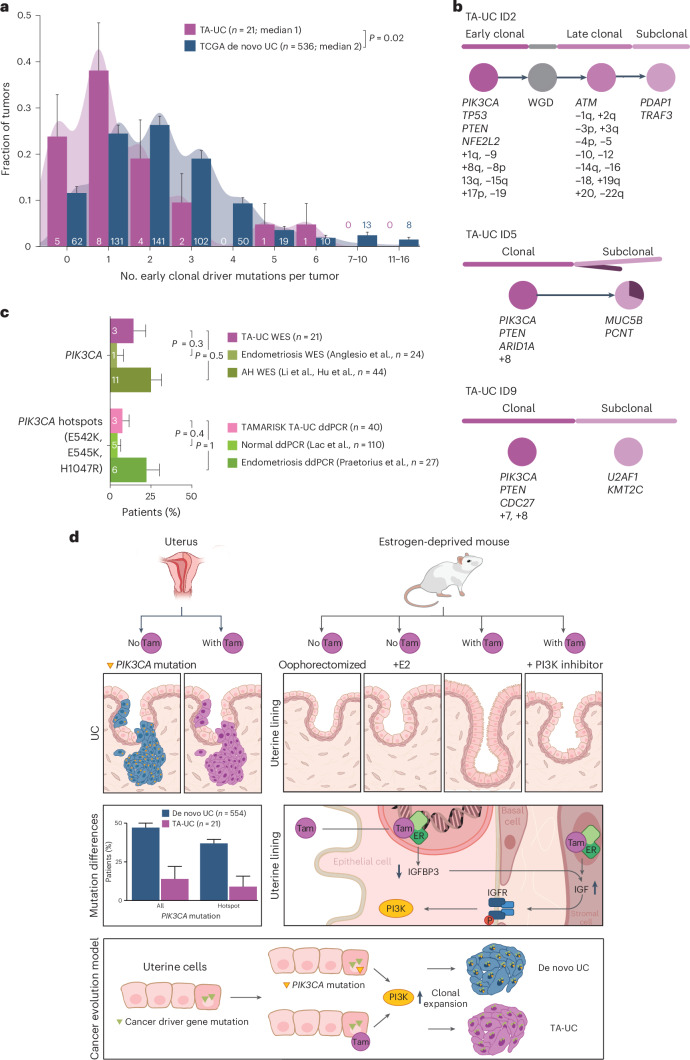
Mutagenic processes and clonal selection contribute to the development of therapy-associated secondary neoplasms, a known complication of cancer treatment. The association between tamoxifen therapy and secondary uterine cancers is uncommon but well established; however, the genetic mechanisms underlying tamoxifen-driven tumorigenesis remain unclear. We find that oncogenic PIK3CA mutations, common in spontaneously arising estrogen-associated de novo uterine cancer, are significantly less frequent in tamoxifen-associated tumors. In vivo, tamoxifen-induced estrogen receptor stimulation activates phosphoinositide 3-kinase (PI3K) signaling in normal mouse uterine tissue, potentially eliminating the selective benefit of PI3K-activating mutations in tamoxifen-associated uterine cancer. Together, we present a unique pathway of therapy-associated carcinogenesis in which tamoxifen-induced activation of the PI3K pathway acts as a non-genetic driver event, contributing to the multistep model of uterine carcinogenesis. While this PI3K mechanism is specific to tamoxifen-associated uterine cancer, the concept of treatment-induced signaling events may have broader applicability to other routes of tumorigenesis.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: G.G. receives research funds from IBM, Pharmacyclics–AbbVie, Bayer, Genentech, Calico, Ultima Genomics, Inocras, Google, Kite and Novartis and is also an inventor on patent applications filed by the Broad Institute related to MSMuTect, MSMutSig, POLYSOLVER, SignatureAnalyzer-GPU, MSEye and MinimuMM-seq, and DLBclass. He is a founder of and a consultant to and holds privately held equity in Scorpion Therapeutics; he is also a founder of and holds privately held equity in PreDICTA Biosciences; and he holds privately held equity in Antares Therapeutics. R.J. received research funding from Lilly, Pfizer and Novartis and serves on an advisory board for GE Healthcare and Carrick Therapeutics. Y.E.M. is a consultant in Foresee Genomics. C.P.P. holds stock and other ownership interests in Xsphera Biosciences and receives honoraria from Bio-Rad and consults or advises for DropWorks and Xsphera Biosciences. C.P.P. also has sponsored research agreements with Daiichi Sankyo, Bicycle Therapeutics, Transcenta, Bicara Therapeutics, AstraZeneca, Intellia Therapeutics, Janssen Pharmaceuticals and Array BioPharma. W.J.G. is a cofounder of and holds equity in Ampressa Therapeutics, is a consultant for and holds equity in inference and has received consulting fees from Boston Clinical Research Institute, Belharra Therapeutics, Faze Medicine and ImmPACT Bio. E.M.V.A. reports an advisory role and/or consulting with Tango Therapeutics, Genome Medical, Invitae, Enara Bio, Janssen, Manifold Bio and Monte Rosa; research support from Novartis and BMS; equity in Tango Therapeutics, Genome Medical, Syapse, Enara Bio, Manifold Bio, Microsoft and Monte Rosa; travel reimbursement from Roche–Genentech and institutional patents filed on chromatin mutations and immunotherapy response and methods for clinical interpretation. W.Z. receives research funding and advises for Astellas Pharma. U.A.M. reports receiving consulting fees received from Merck, Novartis, Blueprint Medicines, AstraZeneca and NextCure as well as participating on a data safety monitoring board or an advisory board for Symphogen and Advaxis. M.J.E.M. receives research funding from W.J. Thijn Stichting. I.L. is a consultant for PACT Pharma and is a board member, scientific advisor and consultant to Ennov1. A.N. is currently employed by AstraZeneca. J.G. and M.R. disclose a financial association with Caris Life Sciences, including full-time employment, travel and/or speaking expenses and stock and/or stock options. The other authors declare no competing interests.
Figures











References
-
- Carthew, P. et al. DNA damage as assessed by 32P-postlabelling in three rat strains exposed to dietary tamoxifen: the relationship between cell proliferation and liver tumour formation. Carcinogenesis16, 1299–1304 (1995). - PubMed
-
- Carthew, P. et al. Cumulative exposure to tamoxifen: DNA adducts and liver cancer in the rat. Arch. Toxicol.75, 375–380 (2001). - PubMed
-
- Busch, H. Adducts and tamoxifen. Semin. Oncol.24, S1-98–S1-104 (1997). - PubMed
MeSH terms
Substances
Grants and funding
- P50CA240243/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- W81XWH-17-1-0084/United States Department of Defense | United States Army | Army Medical Command | Congressionally Directed Medical Research Programs (CDMRP)
- W81XWH-17-1-0084/U.S. Department of Defense (United States Department of Defense)
- K08 CA191058/CA/NCI NIH HHS/United States
- R01 CA237414/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
