

Time-deterministic cryo-optical microscopy
- PMID: 40846863
- PMCID: PMC12373960
- DOI: 10.1038/s41377-025-01941-8
Time-deterministic cryo-optical microscopy
Abstract
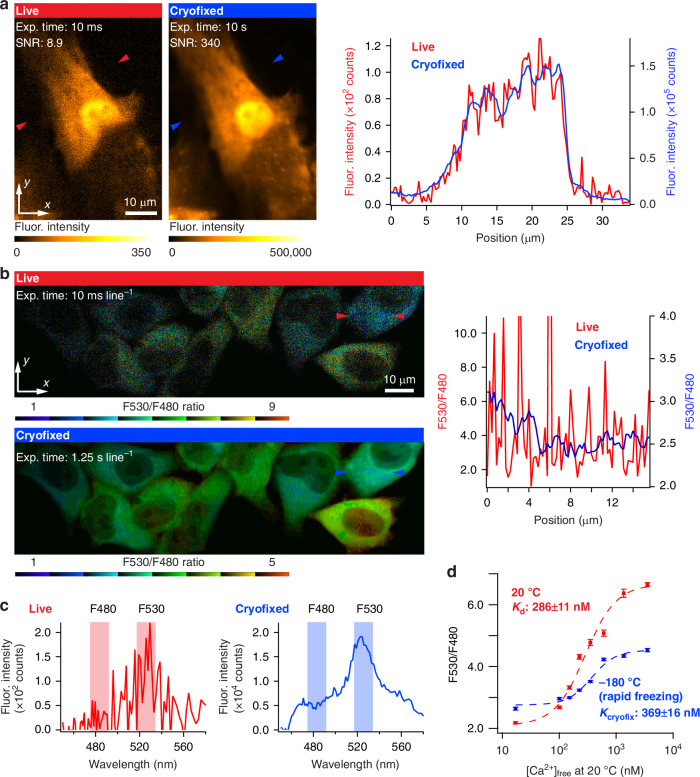
Fluorescence microscopy enables the visualization of cellular morphology, molecular distribution, ion distribution, and their dynamic behaviors during biological processes. Enhancing the signal-to-noise ratio (SNR) in fluorescence imaging improves the quantification accuracy and spatial resolution; however, achieving high SNR at fast image acquisition rates, which is often required to observe cellular dynamics, still remains a challenge. In this study, we developed a technique to rapidly freeze biological cells in milliseconds during optical microscopy observation. Compared to chemical fixation, rapid freezing provides rapid immobilization of samples while more effectively preserving the morphology and conditions of cells. This technique combines the advantages of both live-cell and cryofixation microscopy, i.e., temporal dynamics and high SNR snapshots of selected moments, and is demonstrated by fluorescence and Raman microscopy with high spatial resolution and quantification under low temperature conditions. Furthermore, we also demonstrated that intracellular calcium dynamics can be frozen rapidly and visualized using fluorescent ion indicators, suggesting that ion distribution and conformation of the probe molecules can be fixed both spatially and temporally. These results confirmed that our technique can time-deterministically suspend and visualize cellular dynamics while preserving molecular and ionic states, indicating the potential to provide detailed insights into sample dynamics with improved spatial resolution and temporal accuracy in observations.
© 2025. The Author(s).
Conflict of interest statement
Conflict of interest: K.F. and Y.K. are inventors on the patent related to this work (application number: PCT/JP2022/020045, applicant: The University of Osaka, publication date: 17 November 2022, publication number: WO 2022/239830 A1), and PCT national phase applications have been filed in the United States, Japan, Europe, and China. K.F., M.Y., Y.K., and K.T. have patent applications (application number: PCT/JP2023/038854, applicant: The University of Osaka). K.F. is a co-founder and the chief technology officer (CTO) of Millde Co., Ltd. The authors declare no additional conflict of interest.
Figures



References
-
- Lou, Z. R., Li, P. & Han, K. L. Redox-responsive fluorescent probes with different design strategies. Acc. Chem. Res.48, 1358–1368 (2015). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
